
Iron is a trace element that is essential for life, being required for important cell processes such as DNA synthesis, energy production and defense. Many different structural and enzymatic proteins contain iron, which is essential to their function. In some of these proteins, iron (in the ferrous or Fe2+ form) is found in the center of a porphyrin ring, forming a heme prosthetic group. Notable heme-containing proteins are hemoglobin and myoglobin, which serve as oxygen carriers in blood and muscle, respectively, and the cytochrome P450 superfamily and catalase, which metabolize drugs and degrade hydrogen peroxide, respectively.
Iron is measured in serum or plasma, where it is most commonly used as a marker of iron status (deficiency or excess) and inflammation. Iron in serum or plasma is not “free” (iron is a strong free radical and highly toxic) but is bound to the iron transport protein, apotransferrin (the combination of iron + apotransferrin is called transferrin), in ferric form (Fe3+). Cells acquire iron from transferrin, which binds to transferrin receptors and is taken into the cell through clathrin-mediated endocytosis. Once within the cell, the iron is liberated from transferrin within the acidic compartment of the lysosome. The liberated iron can then be incorporated into proteins or stored as ferritin (iron + the protein apoferritin) or, when in excess, hemosiderin (iron complexed to phosphate or hydroxide). These stores can then be used by the cell for its essential functions if serum iron decreases (ferritin can be more rapidly mobilized than hemosiderin, which is considered “less available” for immedate release to transferrin). Rapidly dividing cells and red blood cell precursors have a high iron requirement (for cell division and hemoglobin synthesis, respectively) and express high concentrations of transferrin receptor. Indeed, immunologic detection of the transferrin receptor is used to identify erythroid progenitors in bone marrow aspirates of humans and rodents.
Iron is conserved within the body. The typical adult human body contains about 3000-4000 mg of iron. Very small amounts of iron are lost from the body per day (1-2 mg, usually through the intestine from sloughed mucosal epithelial cells) and must be replaced through dietary intake (average of 1-2 mg of iron is absorbed from the diet). If dietary iron is inadequate to replace these losses (e.g. vegetarian diet), iron deficiency can ensue – but only once body stores are depleted.
Iron absorption
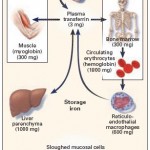
Dietary iron is obtained either from inorganic sources (ferric iron) or animal sources (from breakdown of the heme group in hemoglobin or myoglobin, i.e. blood or muscle). Iron from these two sources are taken up by different transporters or receptors on the luminal surface of enterocytes. Once within intestinal cells, it is used for cellular processes, stored as ferritin (which results in iron loss when the cells slough into feces) or transported out of the cell through the iron transport protein, ferroportin-1, into plasma where it binds to transferrin. The transferrin then delivers the iron to other cells in the body. Intracellular iron is usually in the ferrous (reduced, Fe2+) form and must be converted to the ferric (oxidized, Fe3+) form before it can bind to transferrin. This is accomplished by a copper-dependent enzyme called hephaestin, which is located at the basolateral surface of the enterocyte.
Iron transport
Iron circulates in plasma bound to transferrin (which can bind one or two ferric iron molecules). Any cell can take up iron from transferrin, but by far, erythroid progenitors have the greatest need for iron, which is required for hemoglobin production. In fact, red blood cells contain most of the iron in the body (1800 mg), followed by the liver (as ferritin), macrophages (which recycle hemoglobin iron and store extra as ferritin or hemosiderin) and muscle (as myoglobin) (see image above).
Iron recycling
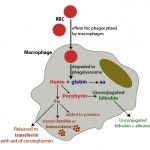
The iron within red blood cells is being turned over constantly. Most of the iron used for red blood cell hemoglobin production is obtained from hemoglobin breakdown of senescent or aged red blood cells (called recycling). Aged red blood cells are phagocytized by macrophages (primarily in the spleen, liver and bone marrow). Once within the macrophage, hemoglobin is broken down by hydrolytic enzymes in the phagolysosome, releasing the heme group (porphyrin ring + iron) and the globin (protein) chains (see image to the right). Globin chains are broken down into their constituent amino acids, which are then used for protein synthesis. Heme oxygenase in the endoplasmic reticulum converts the porphyrin ring within the heme group to unconjugated bilirubin (which is released from the cell into plasma where it binds to albumin and is eventually taken up by the liver) and releases the iron. Similar to the enterocyte, the iron is then transported through ferroportin-1 to transferrin or stored within the cell (as ferritin or hemosiderin). Instead of hephaestin, copper-dependent ceruloplasmin oxidizes macrophage iron so that it can bind to transferrin. Since copper is important for oxidizing iron so it can be transferred to apotransferrin from cells (enterocytes or macrophages), copper deficiency can result in iron deficiency (since iron will not be absorbed or released from stores in macrophages).
Iron uptake
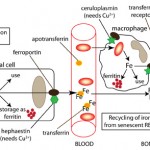
In the bone marrow and spleen, erythroid progenitors usually cluster around macrophages in so-called erythroblastic islands, so that they can immediately bind transferrin that has taken up the iron released by the macrophage and use the iron for hemoglobin synthesis within mitochondria. This uptake of iron through erythrophagocytosis with subsequent release of iron to erythroid progenitors results in rapid turnover of iron within the body.
Iron regulation
Since free or excess iron is detrimental (iron is a free radical promoting oxidant injury), iron is regulated closely in the body. This regulation occurs primarily at the level of the enterocyte, which blocks iron absorption when iron is in excess (so-called mucosal block). There is no regulated pathway for active excretion of iron (can only occur by bleeding or sloughing of iron-laden enterocytes). This block is mediated by a protein called hepcidin, which is produced by the liver. Hepcidin decreases iron absorption in the intestine and release from body stores by causing internalization and degradation of the iron transporter, ferroportin-1, in enterocytes, macrophages and hepatocytes (iron is “sequestered” within these cells). Lack of ferroportin decreases transfer of iron from the diet or body stores to transferrin and results in a rapid decrease in serum or plasma iron concentrations. Hepcidin production is stimulated by bone morphogenic protein (BMP2 and 6) release from liver sinusoidal endothelial cells, which activate the SMAD signaling pathway in erythrocytes (Pagani et al 2019).
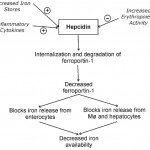
Hepcidin is regulated by the following:
- Available iron stores: Iron stores indirectly (via other proteins, such as transferrin receptor 2) regulate hepcidin concentrations by affecting hepcidin gene synthesis. Increased body iron stores would induce hepcidin gene synthesis, which would then decrease intestinal iron absorption. Note that transient increases in plasma iron (e.g. after oral absorption of ferrous sulfate tablets) will result in hepcidin release, which then limits iron absorption from the intestine for 48 hours (thereby offsetting the benefit of daily oral iron supplementation) (Moretti et al 2015). Decreased body iron inhibits hepcidin release by suppressing secretion of BMP6 from liver sinusoidal endothelial cells. In addition, the BMP receptor is cleaved by increased activity of a protease (TMPRSS6), reducing activation of the SMAD pathway, and there is decreased synthesis of hepcidin mRNA via histone deacetylase3 (Pagani et al 2019).
- Erythropoietic activity: Erythroblasts secrete a hepcidin regulating hormone, called erythroferrone, under the influence of erythropoietin (Kautz et al 2014). Erythroferrone inhibits BMP2/6 release by hepatic sinusoidal endothelial cells (Pagani et al 2019). Erythropoietic activity over-rides any effect of iron stores have on synthesis of this protein. Increased erythropoietic activity inhibits hepcidin synthesis, which makes sense, because erythroid precursors need to obtain iron from the intestine or stores within macrophages for hemoglobin synthesis. However, this still occurs in states of iron excess.
- Inflammatory cytokines: This appears to over-ride both the iron store- and erythropoiesis-sensing regulators of hepcidin. Inflammatory cytokines, particularly interleukin (IL)-6 and tumor necrosis factor (TNF)-α, stimulate hepcidin synthesis via a different pathway than BMP2/6 (STAT3 signaling) (Pagani et al 2019), resulting in decreased iron absorption from the intestine and release from macrophages.Thus, serum or plasma iron concentrations drop rapidly (within a few hours) in response to inflammation, which is why iron can be a serum marker of inflammation. Iron and inflammatory cytokines regulate signaling via the bone morphogenic protein/SMAD. The latter signaling pathway is inhibited by a chaperone or immunophilin, called FKB12, which is a newly discovered intracellular inhibitor of hepcidin (Colucci et al 2017).
Sequestration of iron will limit the amount of iron available for erythropoiesis, which decreases red blood cell production, resulting in the anemia of inflammatory disease, a mild to moderate non-regenerative normocytic normochromic anemia that occurs in inflammatory or “chronic” disease states (note that even though hepcidin is now thought to be a key mediator of the anemia of inflammatory disease, increased hepcidin with iron sequestration is not the only mechanism for this type of anemia; other factors, such as suppression of erythropoietin release, are contributory). The related image shows regulation of hepcidin levels, with subsequent downstream effects on iron in the above scenarios.