
The kidney has many roles, including:
- Elimination of metabolic waste: Function of the glomeruli
- Fluid, electrolyte and acid-base balance: Function of the tubules.
- Conservation of nutrients: Glucose, amino acids – function of tubules
- Glucose: This is freely filtered through an intact glomerulus and resorbed in the proximal convoluted tubules (PCT) by Na-dependent carrier transport mechanisms (sodium-glucose cotransporters or SGLTs). SGLT1 and SGLT2 are found on the luminal side of the renal tubular epithelial cell and are the main glucose transporters in the proximal tubules, carrying around 10 and 90% of the filtered glucose, respectively (Szablewski 2017). These transporters do have set capacities for uptake of filtered glucose, which is species dependent. If the amount of filtered glucose exceeds the transport capacity (called the renal threshold), glucosuria can result. Defective tubular function from proximal tubular disease can result in glucosuria without hyperglycemia. The renal threshold differs amongst species (for values see chemical constituent page). Other transporters, such as GLUT2, transfer glucose from the renal tubular cell into blood on the basolateral side of the membrane. Other transporters will take up D-galactose and fructose (Szablewski 2017).
- Amino acids: These are filtered and resorbed by the PCT via ion-dependent carriers (Na+, K+, Cl–). Different carriers exist for acidic (lysine, arginine), neutral (isoleucine, phenylalanine) and basic (glycine, glutamine) amino acids. Glutamine is used by the proximal renal tubules to create ammonia, which is important for acid-base balance (see below).
- Endocrine function
- Erythropoietin: Peritubular interstitial cells (? fibroblasts) produce erythropoietin, the major cytokine for erythropoiesis. Erythropoietin stimulates the production of erythrocytes in response to hypoxia. Reduced erythropoietin production occurs in chronic renal disease, resulting in a mild to moderate normocytic normochromic non-regenerative anemia. This anemia is responsive to exogenous supplementation of erythropoietin. Recombinant human erythropoietin can be used, but is antigenic in dogs, cats and horses, producing a pure red cell aplasia. Unfortunately, recombinant canine and feline erythropoietin (although both have been cloned) are commercially unavailable.
- Vitamin D: The active form of vitamin D, 1,25(OH)2D or calcitriol, is produced from 25-hydroxycholecalciferol (calcidiol) by α-1 hydroxylase in proximal renal tubular epithelial cells under the stimulation of parathyroid hormone (PTH). Vitamin D stimulates the intestinal and renal absorption of calcium and phosphate and promotes their release from bone (indirectly by facilitating the action of PTH on bone). Note, this does not occur in horses, who lack the enzyme in their renal tubular cells.
- Prostaglandins: Intra-renal production of prostaglandins is very important for maintaining medullary blood flow. The medulla is the most active part of the renal tubule and adequate blood flow is essential to prevent medullary hypoxia. Prostaglandins (and nitric oxide) are vasodilators that function in this regard. Administration of non-steroidal anti-inflammatory drugs will inhibit renal prostaglandin production, predisposing the kidney to medullary hypoxia (and consequent renal failure).
- Renin: This is produced by the juxtaglomerular complex in afferent arterioles of the kidney. The main stimuli for renin secretion are renal hypoperfusion (e.g. hypovolemia) and decreased chloride delivery. Hypoperfusion is sensed by baroreceptors in the wall of afferent arterioles, heart and aorta. The macula densa is a group of cells located at the end of the thick ascending limb of the loop of Henle. In this limb, NaCl is absorbed by the NaK2CL (NKCC2) apical transporter (lumen side). If less NaCl is absorbed by these cells (say due to decreased chloride in the filtrate), renin secretion is stimulated. Renin secretion can also be stimulated by a second “salt sensing” transporter, the apical Na/HCO3– exchanger. Once secreted, renin produces angiotensin I which is converted to angiotensin II (by angiotensin converting enzyme, found mostly in the lungs). Atrial natriuretic peptide inhibits renin (causing an increase in glomerular filtration rate and natriuresis).
- Angiotensin II has two major actions:
- It raises blood pressure by vasoconstriction;
- Promotes sodium and water retention (and thus expands plasma volume) by the kidneys, both directly (stimulates sodium absorption through activation of the luminal sodium/hydrogen antiporter) and indirectly by stimulating secretion of aldosterone from the adrenal cortex. Angiotension II also promotes acid excretion by stimulating sodium absorption (hydrogen is excreted into the urine in exchange for sodium, generating bicarbonate in the process) and bicarbonate absorption (by stimulating the sodium/bicarbonate cotransporter, which pumps bicarbonate and sodium back into interstitial fluid on the basolateral side of the tubular cell; see diagrams under acid-base below).
- Angiotensin II has two major actions:
- Aldosterone promotes sodium retention (and potassium excretion) in principal cells (by stimulating the apical sodium transporter) in the distal tubules of the kidneys, enhancing water retention. Aldosterone also promotes acid excretion by stimulating a luminal hydrogen ATPase pump in type A intercalated cells in the collecting tubules and ducts as well as a chloride/bicarbonate exchanger in the basolateral membrane of the cells (this exchanger pumps out bicarbonate generated within the tubular cells, via carbonic anhydrase, during acid excretion; see diagrams under acid-base below). Aldosterone is stimulated by angiotensin II (in response to hypovolemia, hypo-osmolality, low NaCl [latter sensed by the macula densa]), hyperkalemia and ACTH. It is inhibited by dopamine and atrial natriutic peptide, which causes sodium loss.
Elimination of nitrogenous waste
Most nitrogenous waste is formed by protein catabolism, which is excreted into the urine via glomerular filtration (urea and creatinine are both freely filtered through an intact glomerulus). Amino acids are converted to urea through the urea cycle in the liver. Urea is the main mechanism by which mammalian species excrete nitrogenous waste (uric acid is the main excretory product in birds and reptiles), being produced from ammonia in the liver.
- Urea: Urea is readily filtered by the glomerulus. It is resorbed by the proximal convoluted tubules; resorption being dependent on the flow rate through the renal tubules. Increased flow rate (e.g. polyuria) will decrease urea concentrations in plasma by promoting excretion. Decreased flow rate (due to decreased glomerular filtration, e.g. secondary to hypovolemia) will increase plasma urea by facilitating absorption. Antidiuretic hormone also promotes urea absorption in the collecting tubules. Increased concentrations of urea, measured as [blood] urea nitrogen or [B]UN are seen in the following conditions:
- Decreased GFR from any cause
- Protein catabolism – fever, corticosteroids
- Increased protein digestion – increased dietary intake of protein or hemorrhage into the gastrointestinal tract
- Creatinine: Creatinine is produced in muscle from creatine (which is produced in the liver). Creatinine is freely filtered by the glomerulus and not resorbed. Minimal tubular secretion of creatinine occurs in domestic animals, except for the goat, in which tubular secretion of creatinine can be substantial. Increased concentrations of creatinine occur with decreased GFR. It is a more reliable indicator of GFR than urea as the plasma concentration is less influenced by extra-renal factors.
Electrolyte balance
The kidney plays major roles in maintenance of electrolyte concentrations. Sodium homeostasis is inextricably linked to water homeostasis.
Sodium
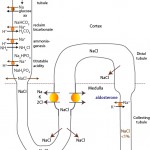
Sodium is filtered and passively resorbed with water along a concentration gradient in the proximal convoluted tubule. The concentration gradient is established by a basolateral Na+/K+-ATPase pump which pumps 3Na+ out of the renal tubule cell in exchange for 2K+. In the PCT, Na+ is cotransported with glucose, amino acids, bicarbonate (NHE3 trasnporter) and phosphate (called titratable acidity). Sodium absorption with bicarbonate retains >80% of all the filtered bicarbonate (with the rest resorbed lower down in the kidney). Sodium absorption concurrently promotes acid excretion in the proximal tubule (and bicarbonate retention; see diagrams under acid-base below). In the thick ascending limb of the loop of Henle, a Na-K-2Cl carrier transports Na+ without water into the medullary interstitium. This is the primary mechanism for establishment of the medullary concentration gradient, which is essential for water resorption. Sodium not absorbed in the loop of Henle is delivered to the distal nephron, where it is also absorbed in the early distal tubule (via a sodium chloride cotransporter called NCC) and then, what is termed the “aldosterone-sensitive distal nephron”, in the late distal tubule and collecting tubules and ducts. In the collecting tubules and ducts, Na resorption occurs via 3 mechanisms: a) A sodium transporter (ENaC) in the apical membranes of principal cells, which is linked to the exchange of potassium in the late distal tubule; b) The sodium transporter in principal cells, but here Na absorption is linked to paracellular Cl absorption (maintaining electroneutrality); and c) With chloride with a pendrin transporter-containing intercalated cells. The last two mechanisms will spare concurrent potassium excretion.
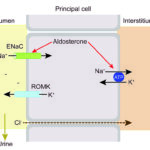
The sodium transporter’s ENaC activity is increased by:
- Aldosterone, which increases Na absorption and promotes K and hydrogen excretion (the latter when potassium is depleted).
- Flow rate in the lumen of the distal nephron, which is largely governed by sodium concentration in the lumen – more distal sodium delivery will prompt more sodium resorption via all three pathways above, with hydrogen or potassium excretion. Distal sodium delivery is increased when there is decreased absorption in the loop of Henle by the Na-K-2Cl carrier (hypochloremia, hypokalemia) or in early distal tubule by the NaCl cotransporters (decreased chloride).
- Bicarbonate: Excess bicarbonate in interstitial fluid (from an alkalemia) or interstitial fluid (filtered when there is a metabolic alkalosis) will stimulate the pump activity.
Chloride
Chloride resorption is both active and passive and both are indirectly linked to Na+ absorption (see above diagram for details). Some active absorption of Cl– (in exchange for formate) occurs in the proximal tubules, which is driven by the Na+/H+ antiporter at the tubule lumen. Passive absorption of chloride occurs along a concentration gradient established by sodium absorption without chloride in the first part of the proximal tubule. The early PCT is relatively impermeable to chloride, increasing the intraluminal Cl– concentration. Cl– is passively absorbed, with sodium, through leaky tight junctions in the later segments of the proximal tubule along a concentration gradient. Also, absorption of water in the proximal tubule causes passive Cl absorption via solvent drag (through the permeable tight junctions). Chloride is also actively resorbed in the loop of Henle with the Na-K-2Cl carrier (i.e. two chlorides are brought in with one potassium and one sodium, which is why loop diuretics can cause a metabolic alkalosis through loss of chloride). Chloride is also absorbed with sodium in the early part of the distal tubules (NCC carrier) and in exchange for bicarbonate in type B intercalated cells in the collecting duct (via pendrin, a chloride/bicarbonate exchanger, which is found at the apical or luminal aspect of the membrane). As indicated above, chloride is also absorbed with the sodium transporter, ENaC, in the late distal tubule through a paracellular mechanism involving claudin-4 and claudin-8.
Chloride delivery to the macula densa cells in the distal tubules is important for control of sodium resorption. Decreased chloride delivery (e.g. hypochloremia) results in reduced NaCl absorption in the macula densa and stimulates the renin-angiotensin-aldosterone system, which promotes NaCl (angiotensin II stimulates the sodium/hydrogen antiporter in the proximal tubules and aldosterone opens sodium channels in the distal tubule) and water resorption (which follows sodium). Loss of chloride with loop (which block the NaK2Cl transporter) or thiazide (block the NaCl transporter in the early distal tubule) diuretics may result in a primary metabolic alkalosis by increasing distal sodium delivery to the collecting tubules and ducts, so it is absorbed by the sodium transporter in the principal cells, thus stimulating hydrogen (and potassium excretion). Again, chloride is the best treatment for an alkalosis induced by loop diuretics.
Potassium
Two-thirds of filtered potassium (K+) is resorbed in the PCT. Some is absorbed in the loop of Henle with NaCl by the Na-K-2Cl carrier. In the DCT and collecting ducts, K+ is excreted passively. In the late distal tubule and collecting tubules, K is also excreted actively through potassium channels (renal outer medullary potassium channel or ROMK and maxi-K channels, the latter of which are triggered by increased distal tubular lumen flow rate) by principal cells. The ROMK channels excrete potassium in exchange for sodium in the collecting tubules because sodium resorption through ENaC by the principal cells creates a negative lumen potential, favoring potassium excretion down the electrochemical gradient (even if potassium is already depleted). Thus sodium resorption with aldosterone seems to be the main driving force for potassium excretion at this site. The excretion of K+ is affected by the following:
- Aldosterone: This enhances excretion in the collecting tubules by stimulating sodium absorption in principal cells in the distal nephron via the sodium transporter (creates a high lumen negative potential difference, i.e. the lumen becomes more negative drawing in a positive, such as potassium into the lumen). Aldosterone is stimulated by acidemia (because it promotes hydrogen loss, see below), hyperkalemia, and the renin-angiotensin II system (which responds to hypovolemia sensed by baroreceptors in the cardiac atria, carotid sinus and afferent arteriole of the kidney and reduced NaCl absorption in the macula densa).
- Tubular flow rate: Increased flow rate (usually due to increased sodium delivery) increases excretion, by allowing less time for absorption.
- Sodium delivery to the distal nephron: Increased sodium delivery increases excretion by increasing tubular flow rate and stimulating the sodium uptake channel in principal cells in the late distal tubule and collecting tubules (ENaC), resulting in sodium absorption. The latter increases lumen electronegativity, so a positively charged K+ can now move out more easily into the lumen and distal flow rates. The activity of the sodium transporter is increased by aldosterone and flow rate, with potassium leaching out into urine via H+/K+-ATPases, ROMK and maxi-K channels at the apical membrane of principal cells.
- Acid-base status: An acute mineral metabolic acidosis (e.g. administration of ammonium chloride) transiently inhibits excretion, resulting in a mild hyperkalemia. In contrast, chronic metabolic acidosis likely promotes excretion via aldosterone effects. Alkalemia promotes excretion in two ways:
- Hydrogen leaves the cells to enter blood to combat the alkalemia, whereas potassium moves into cells and then into the renal tubular lumen along a favorable concentration gradient
- Alkalemia stimulates the basolateral sodium/potassium pump which pumps potassium into the cell, creating a concentration gradient favoring excretion into the urine.
- Presence of unabsorbable anions in the filtrate: These are drugs, such as carbenicillin, penicillin, and endogenous products, such as ketones. These increase lumen electronegativity which promotes potassium excretion.
Calcium
Only ionized calcium is filtered. Calcium is mostly resorbed passively in the PCT (80-85%) following sodium and water resorption. In the thick ascending limb of the loop of Henle and DCT, parathyroid hormone stimulates calcium resorption. Vitamin D has a minor effect on calcium resorption in the DCT (by increasing calbindins, calcium-binding proteins in the renal cells).
Phosphate
Most of the filtered phosphate (80-95%) is resorbed in the PCT with sodium, which is influenced by the following:
- Hormones
- ↓ resorption: PTH (decreases the number of Ph carriers and rate of resorption), glucocorticoids, phosphatonins (e.g. fibroblast growth factor-23).
- ↑ resorption: Vitamin D, thyroxin, growth hormone, insulin-like growth factor
- Acidosis: Inhibits resorption (decreases the number of carriers)
- Extracellular volume (ECV): Increased ECV inhibits resorption by promoting natriuresis.
Magnesium
80% of magnesium (ionized only) is filtered, most of which is resorbed in the thick ascending limb of the loop of Henle (70%), with lesser amounts in the PCT (20-30%) and DCT (10%). Magnesium resorption is influenced by ADH, PTH (stimulates absorption), glucagon, calcitonin, and β-adrenergics. The exact mechanisms involving magnesium homeostasis are poorly understood.
Water homeostasis
Urine concentration is the formation of urine that is hyperosmotic to plasma (plasma has an osmolality of approximately 280-320 mOsm/kg), by resorption of water in excess of solute. Urine dilution is the formation of urine that is hypo-osmotic (< 300 mOsm/kg) with respect to plasma by excretion of water with retention of solute. Both concentration and dilution require correct functioning of the renal tubules. The ability of the kidney to concentrate or dilute urine is assessed by urine osmolality or specific gravity.
- Urine specific gravity: A measurement of the density of urine compared to pure water and is determined using a refractometer. The USG is influenced by the number of molecules in urine, as well as their molecular weight and size, therefore it only approximates solute concentration.
- Urine osmolality: In contrast to USG, urine osmolality (usually measured by freezing point depression) is a direct measure of the number of molecules in urine and is not influenced by molecular weight or size. This varies between animals depending on hydration and need for the kidney to concentrate, but is typically >800 mOsm/kg in healthy dogs (Heine et al 2004).

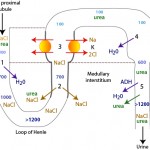
The formation of dilute or concentrated urine is achieved via the countercurrent mechanism which includes the loop of Henle, the cortical and medullary collecting tubules and the blood supply to these segments (vasa recta). Urine concentration or dilution is dependent upon formation of a hypertonic medullary interstitium and the absorption of water in the collecting tubules. Optimal renal concentrating ability is dependent on a number of factors, most important being establishment of the concentration gradient in the interstitium of the renal medulla and antiduiretic hormone (ADH).
- Formation of a hyperosmotic medullary interstitium: This is established primarily by NaCl resorption without water in the ascending limb of the loop of Henle by countercurrent exchange. Urea absorption from the medullary collecting tubule under the action of antidiuretic hormone (ADH) contributes to this process (see image). The medullary concentration gradient is maintained by medullary blood flow in the vasa recta which is arranged in a hairpin configuration to minimize removal of the excess interstitial solute.
- Water absorption: The urine leaving the loop of Henle and entering the collecting tubules is dilute (hypo-osmotic to plasma). To produce a concentrated urine, water must be resorbed. The collecting tubules are normally impermeable to water, however ADH increases their permeability, allowing water to be absorbed along a concentration gradient (established by the countercurrent mechanism) into the medullary interstitium and vasa recta. ADH binds to vasopressin (V2) receptors in the basolateral membrane of principal cells that activate adenylate cyclase and increase intracellular cAMP (Boone and Deen 2008). This then promotes the insertion of water channels (called aquaporins, specifically aquaporin-2) into the luminal membrane of the cell, thus facilitating passive water absorption via a concentration gradient established in the medullary interstitium (via basolateral aquaporins-3 in the cortical and outer medullary collecting duct and basolateral aquaporin-4 in the inner medullary collecting duct). Therefore, production of concentrated urine requires both a hyperosmotic interstitium and ADH. In the absence of ADH or low concentrations of ADH, aquaporin-2 is removed from the luminal membrane by endocytosis, rendering the collecting ducts impermeable to water. If the collecting tubules are impermeable to water, dilute urine is excreted (<50 osM/kg). A dilute urine indicates that the tubules can function to create the dilute urine in the first place, it is just that the tubules for some reason cannot concentrate the urine once it enters the distal/collecting tubules. A less than adequately concentrated urine or hyposthenuric urine can thus be caused by inhibition of ADH action on renal tubules or lack of ADH. ADH is released in response to increases in osmolality (1-3% in dogs) and reductions in effective circulating volume (5-10% in dogs).
Therefore, the tubules have the capacity to dilute AND concentrate urine!! Proximal tubules and loop of Henle create a dilute urine whereas the distal/collecting tubules and ADH create a concentrated urine. Defective renal tubule function with an inability to concentrate or dilute urine will result in isosthenuric urine (or a fixed urine specific gravity of 1.008-1.012), regardless of hydration status. This helps confirm renal disease (due to tubular dysfunction) in an animal. However, factors other than renal tubular disease will alter renal concentrating ability. These factors affect the establishment of the medullary interstitium osmotic gradient (called medullary solute washout) or the ability of ADH to do its job and can result in urine that is inappropriately concentrated (less than “adequate” concentrating ability) in a dehydrated or azotemic animal. In some cases, urine may be hyposthenuric (<1.008). A complete lack of ADH typically results in hyposthenuric urine as the tubules can dilute but not concentrate the urine. These factors are listed below:
- Decreased medullary tonicity (medullary solute washout)
- Sodium absorption: Absorption of NaCl without water in the ascending limb of the ascending loop of Henle is essential for formation of the medullary interstitial osmotic gradient. Hyponatremia (e.g. Addison’s disease) or decreased tubular sodium absorption (e.g. polyuria of any cause which increases tubular flow allowing less time for sodium absorption) will decrease the gradient. The amount of NaCl reaching this part of the tubule is also dependent on GFR and the rate of proximal tubule resorption.
- Medullary blood flow through the vasa recta: Increased blood flow can cause medullary solute washout, e.g. hypokalemia, hypercalcemia, long-standing polyuria
- Urea absorption: Absorption of urea in the collecting tubules, under the influence of ADH, enhances the concentration gradient formed in the medullary interstitium, Decreased urea in the medullary interstitium decreases medullary interstitial osmolality, particularly at the base of the loop of Henle (renal papillary tips). If urea is not being produced (e.g. synthetic liver failure) or absorbed (e.g. polyuria of any cause or lack of ADH), the urine cannot be maximally concentrated.
- Lack of ADH: Lack of ADH production, also called central diabetes insipidus, will result in an inability to concentrate urine, although the urine will be dilute. Suppression of ADH secretion can occur with prednisolone (Bähr et al 2006).
- Inhibition of ADH at the levels of the tubules (e.g. decreased vasopressin receptors) or decreased tubular responsiveness to ADH: This may result in urine that is not adequately concentrated in a dehydrated or azotemic animal or hyposthenuria. This is because ADH is required for insertion of aquaporin water channels that permit water absorption passively from the lumen to the renal interstitium along the concentration gradient in the medulla. Factors which result in this condition (also called nephrogenic diabetes inspidus) are:
- Metabolic alterations: Hypokalemia (downregulates aquaporin-2 [Marples et al 1996]) and free ionized hypercalcemia (various mechanisms proposed, but decreased expression of aquaporin-2 is involved [Khositseth et al 2017]).
- Drugs and hormones: Glucocorticoids (unknown mechanism [Bass et al 1984]) and prostaglandin (E2 and F2α). Prostaglandin E2 inhibits water retention in various ways, but by binding to receptors, it is thought to downregulate increases in cAMP mediated by vasopression. The vasopressin-mediated cAMP increases within the cell induces aquaporin-2 movement to the luminal membrane. Therefore, inhibition of cAMP will result in less aquaporin-2 movement and will decrease collecting duct permeability to water and urea (Olesen and Fenton 2013). Prostaglandin F2α is thought to inhibit the NKCC2 carrier in the ascending limb of the loop of Henle (resulting in salt loss and decreased medullary tonicity) and inhibit the action of ADH on the tubules, resulting in diuresis (Zook and Strandhoy 1981).
- Endotoxin (systemic not just local): Endotoxins downregulate vasopressin receptors and aquaporin-2 translocation to the luminal side of the membrane (Misharin et al 2004).
To summarize:
- An “adequately” concentrated urine (urine specific gravity or USG > 1.030 in the dog, > 1.040 in the cat, > 1.025 in large animals) indicates the renal tubules can dilute and concentrate urine. This makes renal disease unlikely (but does not rule it out) in a dehydrated animal (some animals can have glomerular disease without tubular dysfunction, particularly cats).
- An “inadequately” concentrated urine (USG below the above values but >1.012) in a dehydrated animal indicates impairment of the ability of the tubules to concentrate urine (disease affecting the proximal tubules and loop of Henle) or create a hypertonic medullary interstitium or respond to ADH (without tubular disease) – see factors that affect concentrating ability besides from tubular dysfunction listed above.
- A persistent (fixed) isosthenuric urine (USG 1.008-1.012) in a dehydrated animal indicates that the tubules cannot concentrate or dilute urine. This is usually due to renal disease affecting both proximal and distal tubules.
- A hyosthenuric (dilute) urine (USG <1.008) indicates that the kidney can dilute urine (normal proximal tubule and loop of Henle function) but cannot concentrate urine. This is usually (but not always) due to a lack of ADH.
- Factors which impede establishment of a hypertonic medullary interstitium will affect concentrating ability (see above). If these factors are on board, don’t condemn the kidney out of hand. When these factors are combined with a cause of pre-renal azotemia (eg, hypovolemia due to dehydration), they can mimic findings typical of renal failure, i.e. the urine is less concentrated (or “inadequately” concentrated) than is expected in a pre-renal azotemia (but is usually not isosthenuric or hyposthenuric).
- Although foals have dilute urine after suckling (roughly around 24 hours), they appear to have similar GFR and tubular function to adult animals, and should be able to concentrate when needed (Schott 2011). Similarly calves with diarrhea can increase their urine osmolality and specific gravity (Thornton and English 1976).
For a summary of the players involved in water balance, please go to the fluid changes page under electrolytes.
Acid-base homeostasis
The kidney is essential in maintenance of acid-base balance and a stable pH, which is accomplished by regulating hydrogen excretion so that plasma bicarbonate remains within appropriate limits. Also, changes in renal hydrogen excretion or bicarbonate retention are important compensatory mechanisms for alterations in pH secondary to respiratory or metabolic causes. The kidney can readily handle mild increases in acid or alkali by rapidly excreting one or the other. It is only when the kidney’s ability to excrete acids/alkali is overwhelmed or has not geared up sufficiently that changes in pH occur. Renal control of pH involves two steps:
- Resorption of filtered bicarbonate. Virtually all of the filtered bicarbonate is resorbed, mostly in the proximal tubules (80%), with the rest in the thick ascending limb of the loop of Henle (15%) and around 5% in the cortical and medullary connecting ducts.
- Excretion of the daily acid load produced by amino acid metabolism in the body (titratable acidity). This daily acid load (consists primarily of phosphates and sulfates, but also citrates and hippurates) is quite substantial and if it is not excreted, as in the case of acute kidney injury or chronic renal disease, a titration or high anion gap acidosis can result. In the PCT, resorption of bicarbonate is linked to sodium absorption and H+ excretion (Na/H exchanger or NHE-3), whereas in the collecting tubules, bicarbonate resorption (bicarbonate/chloride exchanger) is linked to the active excretion of H+ via a vacuolar-type or V-H+ATPase or H+/K+ ATPase in type A intercalated cells with chloride following. The H+ATPase requires aldosterone for optimal function and is dependent to some extent on distal flow rate, which is largely determined by distal sodium delivery (concentration). The higher the distal delivery of sodium, the more sodium will be absorbed in this segment (making the lumen more negative) and the more likely that hydrogen will be excreted into the electronegative lumen. Chloride depletion (in metabolic alkalosis) will also promote hydrogen excretion by increasing distal sodium delivery to the collecting tubules and last part of the distal tubule through activating the renin-angiotensin-aldosterone system (RAAS) via reduced ascending limb NaCl absorption. The RAAS is stimulated by reduced NaCl absorption in the macula densa (a group of specialized cells located just after the ascending limb of the loop of Henle in the first part of the distal tubule). The H+/K+ ATPase is stimulated by potassium depletion, in which hydrogen instead of potassium will be excreted, particularly if there is a stimulus for sodium resorption (if a plus goes in, a plus must go out to maintain electroneutrality).
- Compensate for or attempt to correct for acid-base disturbances that are respiratory or metabolic, respectively. This is accomplished via the kidney either generating bicarbonate while simultaneously promoting acid or hydrogen excretion in response to an acidosis or the kidneys retaining acid while excreting or decreasing bicarbonate generation in alkalotic states. Note: The kidney works to try and correct a primary metabolic disturbance (acidosis or alkalosis) when there is normal and optimal renal function but this usually will not normalize the pH, unless the primary disturbance has ceased or is self-limiting (and potentially mild). Normalization of the pH usually requires veterinary (our) intervention to treat and eliminate the condition causing the primary disturbance. Once this condition has been treated and is improving, the acid-base disturbance it is eliciting will start to resolve and then, the kidney’s response will have more meaning and help get the pH back into the normal range (the primary disturbance is no longer driving the pH up or down).
Important concepts relating to the kidney and acid-base balance are:
- Electroneutrality rules! The body (and kidney) attempts to maintain electroneutrality at all costs, i.e. absorption of an anion must occur concurrently with absorption of a cation or in exchange for excretion of an anion (e.g. Na+ absorption is linked to Cl– resorption in the loop of Henle or is absorbed in exchange for H+ excretion in the PCT).
- When an acid is excreted by the kidney, a base is automatically retained in plasma. When a base is excreted by the kidney, an acid is automatically retained in the plasma. So acid excretion is directly linked to base retention and vice versa.
- Bicarbonate and chloride are the only resorbable anions in the filtered fluid or urine within the tubular lumen.
- In acidotic states, the kidney gets rid of excess acid primarily via increasing renal ammoniagenesis (the reverse occurs in alkalotic states). Renal ammoniagenesis occurs primarily in the proximal convoluted tubule but impacts the entire nephron and facilitates (indirectly) chloride-containing acid excretion (as ammonium chloride, NH4Cl) throughout the entire nephron, including the collecting tubules (via increasing ammonia in the interstitium of the kidney; see more below). Thus, both proximal and collecting tubules excrete a chloride-containing acid (ammonium chloride) while retaining or generating base, however the distal tubule is far more efficient at this process. In humans, proximal tubules can acidify urine to a pH of 6.5-6.8, whereas the distal tubules can acidify urine to a pH of 5.0. Dogs and cats cannot acidify urine typically to the latter degree and renal ammoniagenesis appears less efficient in cats versus dogs.
Retention of filtered bicarbonate
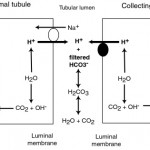
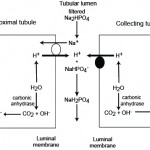
Filtered bicarbonate is retained (“reclaimed”) in proximal and collecting tubules through concurrent excretion of H+ (see figure to the right).
- In the PCT, most of the filtered bicarbonate (around 80% in humans) is resorbed with filtered Na+ in exchange for H+ (via the NHE3 transporter). The hydrogen is derived from carbon dioxide and water through the action of the enzyme, carbonic anhydrase, in the renal tubules, which is tethered to the luminal membrane. The excreted hydrogen combines with filtered bicarbonate and is converted to water and carbon dioxide. The bicarbonate generated within the tubule cell from membrane-bound (CAIV) and cytosolic (CAII) carbonic anhydrase (CA) is transported with sodium into blood through a basolateral (blood side) Na/HCO3 cotransporter. Thus, the actual bicarbonate that is filtered is “lost” in the urine filtrate (by combining with hydrogen to form water and carbon dioxide) but a new bicarbonate is generated by the renal tubule cell during the process of excreting hydrogen (in exchange for absorption of sodium). Thus sodium absorption drives reclamation of filtered bicarbonate. In this way, one bicarbonate is generated for one lost, so there is no net gain of bicarbonate, rather the filtered bicarbonate is being “reclaimed” by the kidney. Angiotension II stimulates the activity of the Na/H antiporter and the basolateral sodium/bicarbonate cotransporter, thus promoting sodium and bicarbonate retention. The energy for the Na/H antiporter actually comes from the Na/K transporter (pumps Na out into blood and K into the tubular cell) in the basolateral (blood side) membrane, so this process is critically dependent on sodium absorption. Bicarbonate is also resorbed in the thick ascending limb of the loop of Henle and collecting tubule (Hamm et al 2015, Dhondup and Qian 2017). There are other transporters for hydrogen, including a luminal H+-ATPase, HNE8, and H/K exchangers, but HNE3 does the most of the heavy lifting with H+ excretion.
- In the collecting tubules, filtered bicarbonate remaining in the urine filtrate is resorbed by hydrogen being actively secreted into the lumen through a luminal vacuolar type (V)-H+ATPase pump via type A intercalated cells. In humans, this equates to about 4% of the bicarbonate (Dhondup and Qian 2017). This pump is normally found in vesicles in the cytosol, which are translocated to the luminal surface. The excreted hydrogen is generated in the tubule cell from CA, which also generates bicarbonate in the process. The bicarbonate is transported into the blood, in exchange for chloride, via a basolateral (blood side) chloride/bicarbonate exchanger (which provides the energy for the translocation of the H+-ATPase). Again, as for the proximal tubules, there is no net gain of bicarbonate in this setting. Aldosterone stimulates the H+-ATPase and the basolateral chloride/bicarbonate exchanger, thus directly promoting acid excretion and retention of base or bicarbonate. The H+-ATPase works differently in the cortical and medullary parts of the collecting tubules. In the cortical collecting tubule, the H+-ATPase is dependent on sodium absorption in adjacent principle cells, thus increased sodium delivery will increase sodium absorption in the collecting tubules by principle cells. This creates a more negative lumen, which favors excretion of a cation such as hydrogen. In contrast, in the medullary collecting tubule, the H+-ATPase activity is sodium independent.
Generation of new bicarbonate
New bicarbonate is generated by the kidney when hydrogen excretion is buffered by anions other than filtered bicarbonate. Thus, when a bicarbonate is generated by the kidney, an acid (hydrogen with different anions, including carbonate, phosphate and chloride) is lost. Similarly, when an acid is not excreted, bicarbonate is “lost” or not reclaimed or generated.
- Phosphate excretion: In the PCT, bicarbonate is generated when Na+ (from filtered sodium phosphate) is absorbed in exchange for H+ (which is produced by carbonic anhydrase in the renal tubular cell). The hydrogen is then excreted in the urine with the filtered phosphate. Thus, sodium absorption and carbonic anhydrase drive new bicarbonate generation, when filtered phosphate (but not bicarbonate) buffers the excreted hydrogen. A similar process occurs in the collecting tubules, although here H+ is actively excreted by the H+ATPase as indicated above. With phosphate filtration, we are not losing bicarbonate (it is not being filtered) so there is a net gain of bicarbonate (the kidney is actually generating new bicarbonate). Phosphate excretion is called titratable acidity and represents the acid produced normally during daily amino acid metabolism and must be removed by the kidneys (to prevent an acidosis). The body has a limited ability to alter the amount of this acid, so only small amounts of bicarbonate are generated by this mechanism (i.e. it is not a way for the kidney to generate bicarbonate [while concurrently losing hydrogen or an acid] to compensate for a primary respiratory acidosis or attempt to correct for a primary metabolic acidosis that is not caused by renal dysfunction). However, the kidney must get rid of the daily acid load (which is substantial) and if it is failing, it cannot do so, resulting in retention of so-called “uremic” acids, which consist of phosphates (primarily), sulfates, hippurates and citrates. Of these, we measure phosphate, which is frequently high in animals with decreased glomerular filtration rate.
-
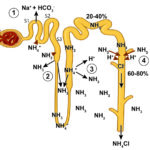
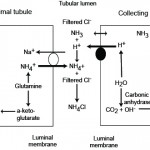
Ammonia metabolism and excretion Renal tubule production of ammonia: The kidney is capable of excreting acid (as ammonium chloride, NH4Cl) and generating new bicarbonate (not just reclaiming filtered bicarbonate), when it produces ammonium (NH4+) from glutamine (an amino acid) in renal ammoniagenesis in the proximal tubules (mostly, accounting for around 70% of ammonia production in basilar states) and loop of Henle.
- S1 and S2 segments of the proximal tubule: Within these upper regions of the proximal convoluted tubular cells, the ammonium is mainly excreted into the tubular lumen. To move into the lumen, the produced ammonium is transported by the apical Na/H transporter (NHE-3) into the tubular lumen in exchange for luminal Na+. Thus, Na+ is absorbed in exchange for NH4+ excretion (not H+ alone). Little ammonia is excreted into the tubular lumen in this segment. Once within the tubular lumen, the ammonium has 2 fates: a) It can combine with chloride to form ammonium chloride (NH4Cl), which is then excreted; or b) The ammonium can be reabsorbed in the ascending limb of the loop of Henle via the Na-K-2Cl carrier (NKCC2), instead of K+. For each NH4Cl that is excreted into the lumen, new bicarbonate is generated and reabsorbed back into blood via a sodium/bicarbonate basolateral exchanger. The bicarbonate actually comes from the α-ketoglutarate that is generated from glutamine metabolism by the enzymes, phosphate-dependent glutaminase and glutamate dehydrogenase. The α-ketoglutarate is converted to bicarbonate when it is used to produce energy via the Kreb’s cycle or glucose via gluconeogenesis. Within the tubular cell, the ammonium can be split off into ammonia (NH3) and a free hydrogen. The ammonia thus produced inside the cell transported across the basolateral membrane into the interstitium and blood and very little is moved into the tubular lumen on the apical or luminal surface of the membrane (see image).
Metabolic acidosis primarily stimulates renal ammoniagenesis in these segments of the proximal tubule, being responsible for up to 80% of the ammonia generated under these conditions. - S3 segment of the proximal tubule: This segment can also produce ammonia as well as ammonium because the tubular cell pH is higher than the luminal pH. The ammonia diffuses or is actively transported into the tubular lumen and is not resorbed in the ascending limb of the loop of Henle because this segment is impermeable to ammonia as well as to water, thus it reaches the distal and collecting tubules. The ammonia also diffuses back into the interstitium (and blood) in this segment of the nephron, contributing to the high medullary interstitial concentration of ammonia (which facilitates distal tubular secretion of acid as ammonium chloride). The ammonium produced in this segment has the same fate as that produced in the S1 and S2 segments (see image).
- Thick ascending limb of the loop of Henle: The ammonium reabsorbed in this segment via the NKCC-2 carrier is moved from the cell across the basolateral membrane into the interstitium via sodium exchange, where it forms ammonia contributing to the high interstitial ammonia concentration. The ammonia also gets back into blood, where it is metabolized in the liver to urea. The absorption of ammonium is quite efficient, removing 60-80% of the ammonia that is normally excreted in urine (as ammonium chloride). Most of the ammonia in the urine then comes from the medullary interstitium via the collecting tubules as outlined below.
- Distal tubules: These do excrete a small amount of ammonia into the tubular lumen.
- Collecting ducts: These segments do not produce ammonium apically like the proximal tubules. Rather, they receive ammonia that was generated by the S3 segment of the proximal tubules and also produce small amounts of ammonia, which can diffuse or be actively transported into the tubular lumen. However, much of the ammonia within the lumen actually comes from interstitial ammonia (which is in higher concentration in the interstitium than the lumen), which diffuses or is actively moved across the cell into the tubular lumen, which has a lower pH than the tubular cell. Once within these segments, the ammonia is trapped as ammonium or NH4+. The H+ that combines with the ammonia to form ammonium comes from the V-H+-antiporter in type A intercalated cells or via H+/K+ exchange. The NH4+ then combines with filtered chloride (or chloride that passively follows H+ excretion via a paracellular pathway) and is excreted. The bicarbonate is moved back into blood via chloride exchange at the basolateral membrane (this is unlike the proximal tubules which use Na+/HCO3– exchange basolaterally). Thus, in the collecting tubules, ammonium is excreted in the urine (accounting for most of the ammonia in urine) and not reclaimed as it is when it is generated in the proximal tubules and reabsorbed in the loop of Henle. This represents a major way for the kidney to get rid of hydrogen as ammonium chloride, however most of the ammonia in this segment is coming from the renal interstitium, which was generated by renal ammoniagenesis in the proximal tubule. In other words, the proximal tubule generates the ammonia the distal tubule uses to excrete hydrogen (directly via the S3 segment or indirectly via producing ammonium which is resorbed by the ascending limb of the loop of Henle)..
In both proximal and collecting tubules, excretion of H+ is linked to generation of new bicarbonate. - Using renal ammoniagenesis, the kidney responds to acid-base disturbances. Alkalemia and hyperkalemia inhibit ammoniagenesis (decreasing acid excretion and promoting bicarbonate excretion), whereas acidemia and hypokalemia stimulate ammoniagenesis (promoting chloride-containing acid excretion and bicarbonate retention). This is summarized as follows:
- To compensate for a primary respiratory acidosis or attempt to correct a primary metabolic acidosis, the kidney must excrete an acid and gain a bicarbonate in the process. This is accomplished by ammoniagenesis with excretion of ammonium chloride (chloride is lost in excess of sodium), which generates a compensating metabolic alkalosis (characterized by a low corrected chloride). In addition, low pH inside the renal tubular cell in response to an acidosis will stimulate translocation of the H+-ATPase from the cytoplasm to the luminal membrane, promoting hydrogen (with chloride passively following via a paracellular) excretion and bicarbonate retention (this will occur more with a respiratory than a metabolic acidosis because, carbon dioxide will easily diffuse across the cell membrane from plasma into the cell and will result in rapid decreases in intracellular pH as the carbon dioxide combines with water to form carbonic acid, generating H+, which stays inside the cell and bicarbonate which is shifted extracellularly in exchange for chloride).
- To compensate for a primary respiratory alkalosis, the kidney decreases ammoniagenesis and ammonium chloride excretion (and the H+-ATPase is not moved to the luminal membrane of type A intercalated cells), thus chloride builds up in the blood and bicarbonate is not generated, creating a secondary or compensatory hyperchloremic metabolic acidosis.
- In a primary metabolic alkalosis due to loss of HCl, the kidney filters the excess bicarbonate and actually actively secretes bicarbonate in the collecting tubules via type B intercalated cells, which flip the basolateral bicarbonate/chloride exchanger, called pendrin, to the luminal membrane so bicarbonate is excreted and chloride is retained (i.e. a hyperchloremic metabolic acidosis). However, the kidney does not do this very well because other factors, hypovolemia, hypochloremia and hypokalemia, impede its ability to excrete bicarbonat. Instead, the kidney excretes acid (and retains bicarbonate), the last thing you want to happen. So instead of fixing the chloride-depleted metabolic alkalosis, the kidney is actually responsible for it (particularly the distal nephron and cortical collecting tubules).
- Concurrent hypovolemia and hypochloremia from the primary cause of the metabolic alkalosis stimulate aldosterone (hypochloremia results in reduced salt sensing in the macula densa) and sodium absorption. Sodium is absorbed with the filtered bicarbonate in the PCT and not with chloride in the loop of Henle (since chloride is also low), which adds the bicarbonate back into blood (the last thing we want the kidney to be doing). The lack of chloride, which is the main problem, increases delivery of sodium to collecting tubule, because less sodium is absorbed via the Na-K-2Cl pump in the loop of Henle and by the NaCl cotransporter in the early distal tubules. Also aldosterone release, stimulated by hypovolemia and hypochloremia, directly stimulates the H+-ATPase in the cortical collecting tubule, further generating more bicarbonate and excreting hydrogen (instead of retaining hydrogen) in the urine, resulting in a paradoxic aciduria. This occurs particularly in cattle with displaced abomasa, abomasal atony or proximal duodenal obstruction, but also has been reported in dogs. Aldosterone worsens this effect by stimulating sodium absorption in principal duct cells, which is linked to hydrogen excretion in the type A intercalated cells in the cortical collecting duct.
- Concurrent hypokalemia (combination of intracellular translocation with alkalemia and losses from the primary cause of the metabolic alkalosis) also exacerbates this situation, since sodium exchanges for hydrogen and not potassium in the aldosterone sensitive portion of the nephron (for more, see below).
To fix this problem and help the kidney do its job to get rid of base or alkali, we administer sodium chloride and supplemental potassium. This restores blood volume and chloride, removing the stimulus for aldosterone secretion (and preventing the kidneys from generating new bicarbonate) and sodium can be resorbed with chloride in latter parts of the proximal convoluted tubule and the loop of Henle (versus with bicarbonate in the PCT) or in exchange for potassium and not hydrogen in the collecting tubules. Thus NaCl is called an acidifying solution (because of its effects on renal excretion of acid/bases) not because it is acidic in itself.
- S1 and S2 segments of the proximal tubule: Within these upper regions of the proximal convoluted tubular cells, the ammonium is mainly excreted into the tubular lumen. To move into the lumen, the produced ammonium is transported by the apical Na/H transporter (NHE-3) into the tubular lumen in exchange for luminal Na+. Thus, Na+ is absorbed in exchange for NH4+ excretion (not H+ alone). Little ammonia is excreted into the tubular lumen in this segment. Once within the tubular lumen, the ammonium has 2 fates: a) It can combine with chloride to form ammonium chloride (NH4Cl), which is then excreted; or b) The ammonium can be reabsorbed in the ascending limb of the loop of Henle via the Na-K-2Cl carrier (NKCC2), instead of K+. For each NH4Cl that is excreted into the lumen, new bicarbonate is generated and reabsorbed back into blood via a sodium/bicarbonate basolateral exchanger. The bicarbonate actually comes from the α-ketoglutarate that is generated from glutamine metabolism by the enzymes, phosphate-dependent glutaminase and glutamate dehydrogenase. The α-ketoglutarate is converted to bicarbonate when it is used to produce energy via the Kreb’s cycle or glucose via gluconeogenesis. Within the tubular cell, the ammonium can be split off into ammonia (NH3) and a free hydrogen. The ammonia thus produced inside the cell transported across the basolateral membrane into the interstitium and blood and very little is moved into the tubular lumen on the apical or luminal surface of the membrane (see image).
Regulation of hydrogen excretion and bicarbonate generation
These are regulated by the following:
- Extracellular pH: Acidemia promotes bicarbonate generation and hydrogen excretion in the tubules, by stimulating ammonia production in the PCT and V-H-ATPase translocation in the type A intercalated cells in the collecting tubule. This likely occurs more with respiratory acidosis causing acidemia, because carbon dioxide diffuses readily intracellularly and these changes require changes in intracellular pH to go into effect. In alkalemia, a subset of intercalated cells (type B) in the collecting tubules actively excrete bicarbonate into the lumen in exchange for chloride by flipping the bicarbonate/chloride transporter (pendrin) from the basolateral membrane to the luminal membrane. Alkalemia can also, adversely, promote the activity of the sodium transporter in the collecting tubules, thus actually promoting acid loss (the last thing you want to be happening in an alkalemia) by the sodium-dependent H+-ATPases in the cortical collecting tubule, when there is increased distal sodium delivery and increased sodium resorption. As indicated above, acidemia and alkalemia also have opposing effects on ammoniagenesis (acidemia increases, alkalemia inhibits).
- Extracellular volume: Bicarbonate is retained (generated) in volume depletion due to decreased bicarbonate filtration and activation of the renin-angiotensin-aldosterone system. In the PCT, angiotensin II enhances H+ excretion (and bicarbonate retention) by increasing the activity of the luminal Na+/H+ antiporter and the basolateral Na+/HCO3– cotransporter (see pictures above). This reclaims filtered bicarbonate and generates new bicarbonate. In the collecting tubules, aldosterone activates medullary and cortical V-H+ATPase and promotes the activity of the basolateral HCO3–/Cl– exchanger, thus enhancing H+ excretion and bicarbonate retention. By stimulating sodium absorption by the sodium transporter in the cortical collecting tubule, aldosterone also indirectly promotes hydrogen excretion by type A intercalated cells, through creating a negative lumen potential after sodium is resorbed.
- Chloride depletion (e.g. vomiting of gastric contents). This produces a metabolic alkalosis (called chloride-depleted metabolic alkalosis) and is the main driver for sustaining the alkalosis by preventing renal acid retention and promoting renal acid excretion.
- In the absence of Cl–, Na+ resorption in the PCT occurs with bicarbonate (also negatively charged) instead of chloride.
- Because the Na-K-2Cl carrier depends on chloride for absorption, lack of chloride in the lumen will decrease sodium absorption in the loop of Henle, resulting in increased delivery of Na+ to the distal nephron, with increased distal tubular flow rates. Decreased sodium chloride absorption (salt sensing) in the macula dense in the first part of the distal tubule will stimulate renin secretion.
- In the early distal tubules, the NaCl transporter will inefficiently absorb the increased sodium presented to it because of the chloride depletion so more sodium gets into the collecting tubules and ducts.
- Once in the collecting tubules, the principal cells absorb the increased amounts of sodium in the tubular fluid via the sodium transporter as they respond to the increased flow rate. Absorption of sodium is “balanced” by excretion of a positively charged molecule, either H+ or K+, by the H+-antiporter or H+/K+ transporters in the type A intercalated cells. The activity of the sodium channel is enhanced by aldosterone, which is stimulated by hypochloremia and hypovolemia, and induces sodium absorption in exchange for potassium excretion and hydrogen secretion by sodium-dependent H+-ATPases in the type A intercalated cells in the collecting tubule. Absorption of sodium is also stimulated by alkalemia and bicarbonate in the tubular fluid. However, potassium is often concurrently low (due to intracellular translocation from concurrent alkalemia and losses from the primary cause of metabolic alkalosis) and cannot be excreted, so Na+ is resorbed in exchange for H+ instead. Furthermore, chloride deficiency will reduce the excretion of bicarbonate by type B intercalated cells in the collecting tubules (because pendrin excretes bicarbonate and does so in exchange for chloride). This creates a paradoxic aciduria, which sustains and potentiates (worsens) a metabolic alkalosis in chloride- and potassium-depleted states.
- Potassium depletion: With hypokalemia, the kidneys try to conserve K+ in exchange for hydrogen, thus promoting acid excretion. Intracellular shifting of hydrogen into cells in exchange for potassium will also increase the amount of bicarbonate in the blood. K+ depletion will also stimulate ammonia production by the proximal tubules (thus generating bicarbonate, see above). Hypokalemia usually occurs as a consequence of metabolic alkalosis and is not usually a primary cause of metabolic alkalosis (unless the hypokalemia is very severe, e.g. primary hyperaldosteronism), although hypokalemia is definitely responsible for worsening or sustaining an alkalosis. A hypokalemia frequently accompanies a metabolic alkalosis because the diseases causing a metabolic alkalosis also result in low potassium (from decreased intake and concurrent losses, as well as potassium moves into cells in exchange for hydrogen in alkalemia). In a primary metabolic alkalosis, hypokalemia exacerbates the alkalosis in all parts of the nephron:
- In the glomeruli, hypokalemia decreases GFR so less bicarbonate is filtered in a metabolic alkalosis.
- In the PCT, hypokalemia promotes renal ammoniagenesis (resulting in additional hydrogen and chloride loss and bicarbonate gain).
- In the loop of Henle, hypokalemia impairs absorption of sodium via the Na-K-2Cl carrier, further promoting hypochloremia (which also stimulates aldosterone after being sensed by the macula densa) and increasing distal delivery of sodium and distal tubular flow rates, which influence sodium absorption in the distal nephron.
- In the early distal tubule, potassium depletion impairs the ability of the tubules to reabsorb sodium with chloride.
- Increased delivered sodium is absorbed in the collecting tubules in exchange for hydrogen instead of potassium under the influence of aldosterone. Hypokalemia also interferes with the ability of ADH to resorb water (downregulates aquaporins), thus exacerbating hypovolemia, which is a stimulus for activation of the renin-angiotensin-aldosterone system.
Therefore animals with metabolic alkalosis due to HCl (vomiting, abomasal atony or outflow obstruction) or KCl loss (sweating in horses) should be treated with chloride (first and foremost, i.e. hypertonic saline) and supplemental potassium.
References
- Clinical Physiology of Acid-Base and Electrolyte Disorders by Rose BD and Post DW, 5th edition, 2001. McGraw-Hill, New York, NY.
- Fluid, Electrolyte and Acid-Base Disorders in Small Animal Practice by DiBartola SP. Elsevier-Saunders, St Louis, MO.
- Gennari 2011. Pathophysiology of Metabolic Alkalosis: A New Classification Based on the Centrality of Stimulated Collecting Duct Ion Transport. Am J Kidney Dis 58:626.
- Rodan 2017. Potassium: Friend or foe? Pediatr Nephrol 32(7): 1109-1121.
- Weiner and Verlander 2013. Renal ammonia metabolism and transport. Compr Physiol 3(1):201-220.