
Bilirubin is considered a test of hepatic function, in essence the ability of the hepatocyte to take up unconjugated bilirubin in blood, conjugate it (render it water-soluble) and excrete bilirubin into bile, where it is broken down in the intestine by bacteria. However, in reality, bilirubin is not used as a test of the functional capacity of the liver (rather bile acids and ammonia are the more common tests used for this) but more as a marker of liver disease (with and without cholestasis) and as supportive evidence for a hemolytic anemia. Although we usually think of bilirubin in terms of its diagnostic utility (i.e. to support a diagnosis of hemolytic anemia or hepatobiliary disease), bilirubin is actually an anti-oxidant, which is its main physiological function.
There are two main forms of bilirubin in blood as determined by the Van den Bergh reactions (reaction with diazo dyes):
- Unconjugated (indirect): This is bound to albumin and is the dominant form of total bilirubin in blood. It is produced in macrophages from breakdown of heme groups (specifically the porphyrin ring of heme). The biggest source of heme is hemoglobin within red blood cells (RBC) but there are other sources, including myoglobin and cytochrome P450 isozymes. Non-RBC sources contribute around 20% of the unconjugated bilirubin. The liver takes up and conjugates the bilirubin (see physiology below). Unconjugated bilirubin has strong hydrogen bonds between its hydrophilic groups, which renders it water-insoluble. Thus, in plasma it is bound to albumin (which is water soluble, allowing unconjugated bilirubin to be transported in plasma).
- Conjugated (direct): Conjugation renders bilirubin water soluble. Only very small amounts in blood because it is normally excreted into bile. It is also the form of bilirubin seen in urine (which is not a normal finding in any species, other than the dog, in which trace to 1+ bilirubinuria may be seen in concentrated urine or a USG > 1.030 [and maybe up to 2+ in highly concentrated urine, > 1.040], and ferret [Orcutt 2003]). Because bilirubin can pass through the glomerular filtration barrier, an increase in conjugated bilirubin in blood will rapidly spill into the urine, thus a bilirubinuria usually precedes a bilirubinemia due to increases in conjugated bilirubin or when there is cholestasis (the exception is delta bilirubin; see below).
Measurement of total bilirubin (direct + indirect) and the bilirubin “split” (direct and indirect bilirubin) in blood (and detection of bilirubin in urine) can be helpful in interpreting changes in test results. For instance, increased breakdown of hemoglobin (e.g. severe hemolytic anemia) will increase the production of unconjugated bilirubin, which is presented to the liver in excess, and can result in increased total bilirubin in blood (mostly unconjugated bilirubin) if the liver’s capacity to take up and conjugate the bilirubin is exceeded. In contrast, an obstruction to bile flow will increase conjugated bilirubin within hepatocytes which will then be refluxed back into blood (and spill into urine), resulting in an increase in total bilirubin, that is mostly due to conjugated (or direct) bilirubin.
Note, that there is a third form of bilirubin, called delta bilirubin (or biliprotein), which is conjugated bilirubin bound to proteins. Delta bilirubin increases in serum when hepatic excretion of conjugated bilirubin is impaired (cholestasis) and the liver retains intact conjugation mechanisms. It has a long half-life (similar to proteins) and is not excreted in the urine (as it is protein bound), requiring removal via general protein catabolism. Delta bilirubin may be responsible for a persistent bilirubinemia without bilirubinuria seen in some animals with cholestasis. It does react with the diazo dyes, similar to direct or conjugated bilirubin, but is usually not provided as part of routine chemistry results.
Physiology
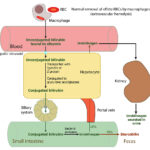
The majority of bilirubin (80%) is produced from the degradation of hemoglobin from erythrocytes undergoing normal (removal of aged or effete cells) or abnormal destruction (i.e. intravascular or extravascular hemolysis) within mononuclear phagocytes (principally splenic, hepatic and bone marrow macrophages). A small percentage (20%) is derived from the catabolism of various hepatic hemoproteins (myoglobin, cytochrome P450) as well as from the overproduction of heme from ineffective erythropoiesis in the bone marrow.
Within macrophages, a free heme group (iron + porphyrin ring) is oxidized by microsomal heme oxygenase into biliverdin and the iron is released (the iron is then stored as ferritin or released into plasma, where it is bound to the transport protein, transferrin). Biliverdin reductase then reduces the green water-soluble biliverdin into unconjugated (water-insoluble but lipophilic) bilirubin. Heme oxygenase is also located in renal and hepatic parenchyma, enabling these tissues to take up heme and convert it to bilirubin. Birds lack biliverdin reductase, thus they excrete heme breakdown products as biliverdin rather than bilirubin.
Unconjugated bilirubin is then released into plasma where it binds to albumin. Uptake of unconjugated bilirubin occurs in the liver and is carrier-mediated and passive. The transport protein is currently unknown, but is generally believed to be organic ion transporting polypeptide (OATP) transporter. This protein, on the sinusoidal membrane of hepatocytes, also transports unconjugated bile acids and dyes such as bromosulphalein (BSP) from the blood into the hepatocyte. Once within the hepatocyte mitochondria, unconjugated bilirubin is transported with ligandin (Y protein or glutathione-S-transferase A) or other proteins (e.g. fatty acid binding protein) and the majority is conjugated to glucuronic acid by UDP-glucuronyl transferase (from the UGT1A1 gene). The remainder is conjugated to a variety of neutral glycosides (glucose, xylose). In the horse, the majority of bilirubin is conjugated to glucose. Bilirubin must be conjugated before it can be excreted into bile (conjugation makes bilirubin water soluble, because it breaks apart the hydrogen bond within the molecule, which keep it hydrophobic and in so doing, makes the central methylene group available to the diazo reagent for measurement). The UDP glucuronyl transferase has been identified in rat and canine kidney, supporting the possibility that canine renal tubular epithelial cells can take up heme, convert it into bilirubin and conjugate it. Conjugation also inhibits binding to albumin and other intracellular proteins. Excretion into biliary canaliculi is the rate-limiting step of the entire bilirubin metabolism pathway and occurs on the canalicular membrane of the cell via a specific transporter, multidrug-resistance associated protein-2 (MRP2), which is energy (ATP) dependent. Transfer into the canaliculi is facilitated by the osmotic force generated by bile salt-dependent and bile salt-independent biliary flow (which is mediated by a chloride-bicarbonate exchanger, which pumps bicarbonate into bile). The force for most of the transporters (on the sinusoidal or canalicular membranes) is generated by a sinusoidal Na+/K+ATPase pump (pumps sodium into the cell and potassium into the extracellular fluid or blood). Bilirubin is transported actively into bile against a concentration gradient (for an image of the bilirubin transporters, refer to the cholestasis page).
Bilirubin is excreted, along with bile salts (and sodium) into the intestine, where bile salts form micelles facilitating absorption of fat. Because conjugated bilirubin is hydrophilic, it cannot be absorbed across the intestinal epithelial barrier. In the intestine, bacterial and intestinal enzymes reduce conjugated bilirubin to urobilinogen. Urobilinogen is re-absorbed (about 10%) or broken down (90%) into urobilin and stercobilin (both of which are excreted in the feces). Of the resorbed urobilinogen, most is taken up by the liver (enterohepatic circulation, i.e. the urobilinogen is absorbed into the portal vein, taken up by the liver and re-excreted into bile – this re-excretion into bile is not depicted in the image above), while the rest bypasses the liver and is excreted into the urine (so small amounts of urobilinogen are found in urine). Bacterial enzymes can also deconjugate bilirubin and the unconjugated bilirubin can be absorbed as well.
Methods
Reaction type
Blanked end-point diazo reaction
Procedure
The method used at Cornell University to measure total bilirubin is the diazo reaction, where a diazo-based dye binds to bilirubin. Only conjugated bilirubin can react with the diazonium ion, because the conformation of unconjugated bilirubin shields or protects the central methylene [CH2] group within the molecule, which is what the diazo dye interacts with. So in order to measure total bilirubin, the sample is treated with a suitable solubilizing agent (usually an alcohol). The addition of an alcohol disrupts the hydrogen bonds that protect the central methylene group, which then reacts with the diazo dye (3,5-dichlorophenyl diazonium), along with conjugated bilirubin, thus measuring total bilirubin in a strongly acidic medium (pH 1-2). The color intensity of the red azobilirubin formed is directly proportional to the total bilirubin and can be measured spectrophotometrically.
Conjugated bilirubin is measured similarly, using a diazo method based on the Jendrassik-Grof procedure, which uses nitrous acid to form the diazonium ion, which binds to conjugated bilirubin in an acidic pH forming azobilirubin, which is red and can be measured spectrophotometrically in an end-point reaction.
Conjugated bilirubin can be measured in the same reaction by omitting the solubilizing agent (since it is already in a conformation where the diazo dye can react – this is because conjugation itself disrupts those hydrogen bonds). Unconjugated bilirubin is then calculated from the formula:
Unconjugated bilirubin = Total bilirubin – conjugated bilirubin
Note, that other analyzers (e.g. dry chemistry analyzers) can measure both delta bilirubin and unconjugated bilirubin directly, but ours cannot.
Units of measurement
The concentration of bilirubin is measured in mg/dL (conventional units) and μmol/L (SI units). The conversion formula is shown below:
mg/dL x 17.10 = μmol/L
Sample considerations
Sample type
Serum, plasma
Anticoagulant
Heparin (lithium, sodium) or EDTA
Stability
The sample must be protected from light, because bilirubin is readily oxidized.
Interferences
- Lipemia, hemolysis: With some analyzers and reagents, hemolysis and lipemia (even mild) will cause falsely high total bilirubin values. The procedures used by the chemistry analyzer at Cornell University are minimally impacted by hemolysis and lipemia.
- Icteric index: The analyzer also gives an estimation of the amount of bilirubin in the sample (free from hemolysis and lipemia interference) as an icteric index. This index correlates closely (often to the nearest 1-2 mg/dL) to the total bilirubin values and can be used to confirm true increases in total bilirubin. Remember that bilirubin is unstable in light and samples stored for several days, in the presence of light, may have falsely reduced bilirubin concentrations. However, in grazing animals, carotenoids also contribute to the icteric index so the index may be a poorer estimate of total bilirubin concentration in these species.
Test interpretation
General interpretation
Clinical icterus is observed when total bilirubin concentrations exceed 3 mg/dL (Joseph and Samant 2021). Note that icterus and cholestasis are not synonymous. Icterus just means a high enough bilirubin to be seen by the naked eye on mucous membranes or in plasma/serum and can result from cholestasis or other causes of hyperbilirubinemia (e.g. hemolytic anemia). You can, of course, have hyperbilirubinemia without visible icterus.
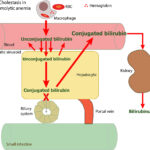
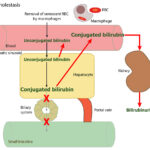
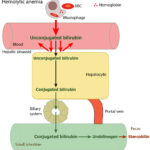
At Cornell University, we interpret changes in total (and direct and indirect) bilirubin concentrations based on pathophysiologic processes which are:
- Increased production of unconjugated bilirubin that is presented to the hepatocyte: The most common cause is hemolytic anemia due to extravascular hemolysis. Small amounts of bilirubin can be produced from intravascular hemolysis as well (see image below), but most of the bilirubin in a hemolytic anemia with an intravascular component is due to the concurrently occurring extravascular hemolysis (this is because most of the free hemoglobin, which is liberated into plasma with intravascular hemolysis, ends up being lost in the urine versus being converted to bilirubin in macrophages). Usually indirect bilirubin dominates (with no bilirubinuria) in hemolytic anemia, particularly when bilirubin concentrations are <3-5 mg/dL, however in some dogs, usually with severe hemolysis (e.g. immune-mediated hemolytic anemia) and higher bilirubin concentrations, cholestasis can develop and dominate, resulting in higher direct versus indirect bilirubin in blood, and concurrent bilirubinuria. The reason why the cholestasis occurs in primarily dogs with hemolytic anemia (but also foals with neonatal isoerythrolysis) is unknown. Prevailing hypotheses are that it is due to a combination of hypoxic injury to hepatocytes with ATP depletion decreasing excretion of conjugated bilirubin by transporters on the canalicular side of the hepatocyte combined with increased production (think of a funnel with unconjugated bilirubin coming into the wide open end of the funnel and a very thin funnel at the end through which conjugated bilirubin leaves and which keeps narrowing over time).
- Defective uptake of unconjugated bilirubin from blood by the hepatocyte: Liver disease or alterations in sinusoidal (blood-side) hepatic transporters taking up unconjugated bilirubin from blood (e.g. cytokines downregulate both the OATP transporter and the Na/K ATPase that provides the energy for the pump). This can result in increased indirect (and total) bilirubin concentrations in blood (but not bilirubinuria). Similarly, defective uptake of unconjugated bilirubin could occur if there is competition for the shared OATP transporter.
- Defective conjugation of unconjugated bilirubin within the hepatocyte: This is usually due to liver disease but could also be due to decreased availability of conjugation compounds. Like decreased uptake, this may increase total bilirubin concentrations, which will be mostly unconjugated. Decreased uptake and decreased conjugation are postulated mechanisms for fasting hyperbilirubinemia in horses (see below) and also likely occur to a lesser extent in cattle with anorexia. The glucuronyl transferase enzyme is not efficient at birth, which may explain higher bilirubin concentrations seen in neonatal foals in the first few days of life (combined with hemolysis of fetal red blood cells) – so-called “hepatic immaturity”.
- Cholestasis or defective excretion of bile: This is defined as reduced or ceased bile flow and can be due to physical or structural impediments to bile flow (structural cholestasis) and altered function of transporters (“functional” cholestasis, e.g. inherited defects in the transporters). Cholestasis will result in bilirubinemia, with increased direct bilirubin concentrations, and bilirubinuria (excess conjugated bilirubin in blood is excreted into the urine, because it is water soluble), with the latter preceding the former. Indirect bilirubin concentrations are also usually increased in cholestasis due to the toxic effect of accumulated bile salts on hepatocytes or cholestasis-induced decreases in the hepatic transporter (OATP), which takes up unconjugated bilirubin from blood. Conjugated bilirubin may also compete with unconjugated bilirubin for uptake by hepatocytes. For more information on these transporters, refer to the cholestasis page.
Note that the following terms are used in some clinical pathologic textbooks to characterize changes in total bilirubin (or icterus): pre-hepatic, hepatic and post-hepatic.
- “Pre-hepatic” hyperbilirubinemia: is due to increased production of bilirubin from breakdown of heme-containing proteins (mostly hemoglobin from RBCs in hemolytic anemia). Here, unconjugated bilirubin is expected to exceed conjugated bilirubin concentrations, which may be within reference intervals. An exception to this is dogs with hemolytic anemia (particularly immune-mediated or IMHA; see more below) and foals with neonatal isoerythrolysis, who develop cholestasis as part of the disease process.
- “Hepatic” hyperbilirubinemia: This is due to defective uptake or conjugation of bilirubin by the hepatocyte and decreased excretion of conjugated bilirubin into biliary canaliculi from structural or “functional” cholestasis. Here, depending on the disorder, unconjugated bilirubin is expected to dominate, with some increases observed in conjugated bilirubin, although if cholestasis is dominating the picture, conjugated bilirubin can be higher than unconjugated bilirubin concentrations.
- “Post-hepatic” or “extrahepatic” hyperbilirubinemia: This is due to problems occurring outside the liver that affect biliary flow within the gall bladder, bile duct, or biliary papilla in duodenum, and is a variant of structural cholestasis. Here, conjugated bilirubin concentration is expected to be increased and usually dominates (and results in accompanying bilirubinuria), with smaller increases in unconjugated bilirubin concentrations. A bilirubinuria will precede a hyperbilirubinemia due to increased conjugated bilirubin.
The bilirubin split
Cornell University is one of the few laboratories that provides results for unconjugated (indirect) and conjugated (direct) bilirubin on our small and large animal chemistry panels. Some clinicians and clinical pathologists do not find the bilirubin split useful because results do not always go cleanly as one would predict from the “pre-hepatic”, “hepatic” and “post-hepatic” classification scheme. This is because in any disease, more than one of the above 4 pathophysiologic processes may be occurring at any given time or may develop over time. For instance, with the first three pathophysiologic processes, unconjugated (indirect) bilirubin frequently exceeds conjugated (direct) bilirubin. However, with hemolytic anemia, particularly in dogs, cholestasis does occur and can dominate the liver biochemical findings (more conjugated than unconjugated bilirubin). In cholestasis, conjugated bilirubin will usually exceed unconjugated bilirubin (except in equidae – see below). However, with decreased bile flow, bile acids accumulate and injure the liver, resulting in increased concentrations of unconjugated bilirubin, which can dominate in some animals. Thus, various combinations of increases in indirect and direct bilirubin may be seen in any one animal. We do find the bilirubin split somewhat useful (which is why it is still included on our chemistry panels), as long as it is interpreted in context of the patient and the rest of the clinical pathologic and other diagnostic testing results.
The bottom line is that if unconjugated bilirubin is higher than conjugated bilirubin, any of the above 4 mechanisms can be operating, but cholestasis is unlikely to be dominating (except in the horse), even though it may be occurring. Rather, increased production, defective uptake and/or conjugation is the dominant process. The results are not specific for any disease, but one should think of the following conditions first (and then find evidence to support their existence): Fasting or anorexia in a horse or ruminant, hemolytic anemia (if evidence of anemia due to hemolysis or reduced red blood cell lifespan) and liver disease. On the other hand, if conjugated bilirubin is higher than unconjugated bilirubin, cholestasis is the dominating pathologic process (since the liver can take up and conjugate the unconjugated bilirubin) and the animal should be examined for causes of cholestasis (intrahepatic and extrahepatic, structural and “functional” not just extrahepatic). Increases in conjugated bilirubin are not specific as to cause, since this can occur secondary to a hemolytic anemia (in dogs with immune-mediated hemolytic anemia and foals with neonatal isoerythrolysis, in particular), parenchymal liver disease that interferes with biliary excretion by affecting bile transporters or physically impeding bile flow, sepsis (inflammatory cytokines impair biliary excretion by downregulating bile transporters and upregulation of nitric oxide secondary to inflammatory cytokines inhibits secretion of bile by biliary ductules), and physical obstructions to bile flow in the biliary system (e.g. cholelithiasis) or too thick bile that does not flow well (bile sludging, e.g. in cats with dehydration). With extrahepatic bile duct obstruction or biliary rupture, levels of conjugated bilirubin are typically higher than unconjugated or indirect bilirubin and produces marked increases in total bilirubin in dogs and cats (20-30 mg/dL). Horses with naturally occurring diseases causing cholestasis are an exception. In this species, direct bilirubin concentrations are increased in cholestasis but are typically lower than the increases in indirect bilirubin concentrations (the direct bilirubin proportion is usually <50% of total bilirubin and often <25-30% of the total bilirubin concentration) (Traub et al 1982, McGorum et al 1999, Peek and Divers 2000, Johnson et al 2006). In contrast, direct bilirubin dominates the total bilirubin split in horses with experimental ligation of the bile duct (Bauer et al 1990).
Urine bilirubin
Total bilirubin concentrations should always be interpreted with knowledge of urine bilirubin, whenever possible. The bilirubin pad on the dipstick only reacts with conjugated bilirubin (no reagent is added to enable the diazonium ion to be added to unconjugated bilirubin). In addition, being water soluble, this is the only form seen in the urine. Increased conjugated bilirubin in blood will produce bilirubinuria (since conjugated bilirubin is small enough to pass through the intact glomerular barrier), which usually precedes the bilirubinemia. In all species, other than the dog and ferret, bilirubinuria is usually diagnostic for cholestasis, since only conjugated bilirubin is present in urine and will spill into urine first before it is increased in blood. In dogs, we need to consider the strength of the dipstick bilirubin reaction in light of the degree of urine concentration to recognize excess bilirubinuria – a trace to 1+ dipstick reaction can be normal in concentrated urine of dogs, particularly males, but is potentially excessive in urine with a USG <1.030 (and likely excessive in a USG <1.015). A 3+ bilirubinuria is not expected in dog urine regardless of how concentrated it is. Note that bilirubinemia can be seen in the absence of bilirubinuria in chronic cholestasis, where delta bilirubin may form (conjugated bilirubin binds to albumin and is not filtered).

Increased concentration of total bilirubin and its components
- Physiologic
- Fasting: In horses, fasting will produce a hyperbilirubinemia due to unconjugated bilirubin. The precise mechanism for this is unknown but it is thought to be due to either decreased uptake of bilirubin (from competition for uptake with free fatty acids) (Naylor et al 1980), changes in conjugating enzymes with fasting, or impaired conjugation of bilirubin (Engelking 1993). The latter has been postulated to be due to low glucose within hepatocytes (in horses most of the unconjugated bilirubin is conjugated to glucose), however after intravenous, intraduodenal or intragastric glucose administration in fasting horses, total bilirubin concentrations were only reduced by 7% after IV dosing in one study (Gronwall and Engelking 1982). Increases in total and indirect bilirubin concentration are noticeable within 12 hours of fasting and may reach levels as high as 10-12 mg/dL within 2-4 days of fasting, with clinical icterus. This occurs in the absence of significant liver disease. Note that horses have higher reference intervals for bilirubin than donkeys. No changes in total bilirubin concentration occur in dogs for up to 8 hours after eating (Yi et al 2022).
- Neonatal: Young animals, especially foals, often have jaundice (due primarily to unconjugated bilirubin). Calves can have mild increases in total bilirubin concentration (usually mostly unconjugated) (up to 1.3 mg/dL) between 1-14 days of age (Lumsden et al 1980, Mohri et al 2007). This is due to multifactorial causes, including hemolysis of fetal red blood cells, decreased liver uptake of bilirubin, immaturity of hepatic conjugation mechanisms and poor albumin binding.
-
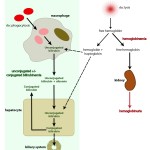
Bilirubin in hemolytic anemia Pathophysiologic
- Anorexia: See above for fasting in horses. Mild increases in total bilirubin (mostly unconjugated) are also seen in cows that are sick and/or anorectic. The total bilirubin usually does not exceed 4 mg/dL in these cows. The precise mechanism is unknown, but postulated to be defective uptake (McSherry et al 1984).
- Hemolytic anemia: Destruction of red blood cells, whether through extravascular or intravascular hemolysis, will increase the production of unconjugated bilirubin because of enhanced hemoglobin metabolism by mononuclear phagocytes (most of the bilirubin in a hemolytic anemia due to intravascular and extravascular hemolysis is from the extravascular component, because the free hemoglobin that is filtered in the urine is “unavailable” for bilirubin production). A healthy liver can handle substantial hemolysis without allowing an increase in total bilirubin, therefore, hyperbilirubinemia is usually due to severe, rapid hemolysis. In these cases, the bilirubin is mostly unconjugated and the total bilirubin is usually <3-5 mg/dL. In some cases of hemolytic anemia (perhaps with longer standing hemolysis), cholestasis ensues and dominates the biochemical results. This could be secondary to hepatic hypoxia/dysfunction will interfere with bilirubin excretion into the bile ducts (remember this is the rate-limiting step of bilirubin metabolism and is ATP-dependent). This occurs predominantly in dogs with immune-mediated hemolytic anemia and some foals with neonatal isoerythrolysis (NI, up to 40-60% of total bilirubin may be conjugated in foals with NI) (Boyle et al 2005). High bilirubin concentrations in foals with NI are associated with the development of kernicterus (bilirubin is neurotoxic) and death (Polkes et al 2008). Therefore, animals with hemolytic anemia and bilirubinemia >5 mg/dL often have a cholestatic component to the icterus, i.e. there are substantial increases in both conjugated (which can dominate) and unconjugated bilirubin, along with bilirubinuria. This reflects both cholestasis and increased unconjugated bilirubin production from hemoglobin breakdown. Note that icterus in cattle is mostly due to hemolysis (and is usually unconjugated) and rarely due to liver disease or post-hepatic bile duct obstruction.
- Liver disease: Hepatic disease may cause increases in both unconjugated and conjugated bilirubin. Increases in bilirubin in the blood of dogs often occurs after increases are seen in the “cholestatic” enzymes (GGT, ALP) due to the low renal threshold for bilirubin. In acutely developing icterus, ALP and GGT activity may be normal because they require time for induction. In large animals with liver disease, increases in bilirubin are usually due to the unconjugated form. Only cattle with severe liver disease will have increased bilirubin (usually unconjugated). Note, that liver disease does not mean the animal is in liver failure. Although increases in bilirubin (likely a mixture of conjugated and unconjugated) are seen in dogs with hepatic failure (Toulza et al 2006) (likely due to a combination of reasons, such as inflammatory cytokine-mediated alteration in transporters, toxicity from accumulated bile acids, alterations in intrahepatic blood flow from fibrosis, hepatocyte swelling resulting in cholestasis), the high bilirubin is not specific for failure and can be seen in various diseases that are not associated with liver failure, e.g. hepatic lipidosis in cats, toxic injury etc. Also note, that a low albumin (as seen with synthetic failure or various other conditions, such as protein-losing enteropathy) resulting in decreased transport of unconjugated bilirubin to the liver is not a cause of high unconjugated bilirubin in animals. There is far more albumin (g/dL) than unconjugated bilirubin (mg/dL) so a low albumin does not impact bilirubin uptake or clearance by the liver.
- Cholestasis: This is defined as decreased or ceased bile flow and can be due to physical obstruction of bile flow or functional or inherited defects in the transporters that deliver bile salts (the bile salt export pump, BSEP) or bilirubin (MRP2) into the biliary system. Obstructed bile flow can be intrahepatic (e.g. hepatocyte swelling due to hepatic lipidosis in cats) or extrahepatic (e.g. bile duct obstruction from pancreatic neoplasia, cholelithiasis, Fasciola hepatica in cattle). Changes in the character of bile (e.g. thick sludged bile in cats with dehydration, mucoceles in dogs) can also result in decreased bile flow. Functional defects in bile salt or bilirubin transporters occur secondary to inflammatory cytokines (e.g. endotoxemia) and drugs. Changes in transporter function also occur with physical obstructions to bile flow (i.e. MRP3 upregulation on the sinusoidal membrane, which pushes bilirubin and bile salts out of the hepatocyte and back into blood, and downregulation of the sinusoidal pumps that shift bilirubin and bile salts into the hepatocyte from blood, Ntcp and OATP).
- Cholestasis will result in bilirubinemia, with increased direct bilirubin, and bilirubinuria (excess conjugated bilirubin in blood is excreted into the urine, because it is water soluble, with bilirubinuria preceding bilirubinemia).
- Indirect bilirubin is also usually increased in cholestasis due to the toxic effect of accumulated bile salts on hepatocytes or cholestasis-induced decreases in the hepatic transporters which take up unconjugated bilirubin from blood.
- Cholestasis frequently (but not always) results in a higher conjugated than unconjugated bilirubin, particularly when there is a physical obstruction to bile flow (e.g. cholelithiasis, biliary mucocele). The exception is the horse, where unconjugated bilirubinemia still dominates in cholestatic conditions, i.e. direct bilirubin rarely exceeds 50% of total bilirubin in horses with cholestasis.
- For more information on expected laboratory findings in cholestasis, refer to the cholestasis page.
- Inherited: Inherited defects in hepatic uptake, conjugation and excretion of bilirubin occur in monkeys, sheep, tamarins and rats. Southdown sheep show defective clearance of bile acids, bilirubin and BSP, supporting a carrier defect in uptake of bilirubin, however they also have increased conjugated bilirubin, suggesting concurrent defects in canalicular transport (Engelking and Gronwall 1979). The syndrome in these sheep is similar to Gilbert’s syndrome in human beings. Corriedale sheep and Golden lion tamarins have Dubin-Johnson syndrome (Engelking and Gronwall 1979) and Golden Lion tamarins (Schulman et al 1993), which is due to defective transport of conjugated bilirubin into bile, resulting in a fasting hyperbilirubinemia, mostly conjugated.