
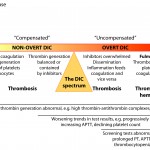
Disseminated intravascular coagulation (DIC) is the abnormal (excessive) activation of hemostasis, with subsequent generation of excess thrombin and formation of microvascular thrombi (arterioles, venules, capillaries). As coagulation factors and platelets are consumed, inhibited or cleaved, hemorrhage can ensue. The disorder is not a simple syndrome, but is rather a continuum of hemostasis activation, beginning with a trigger (initiation phase) that activates coagulation, generating thrombin. Thrombin amplifies its own formation (amplification phase) on the negatively charged surface of cells and cell-derived extracellular vesicles (EVs) bearing phosphatidylserine (PS) or extruded DNA. Initially, thrombin generation is contained or restricted by inhibitors and at this stage, DIC is called “compensated” or is in the “non-overt” stage. The affected animal is considered “hypercoagulable” (defined as excess generation of thrombin) or at risk of thrombosis and may actually be suffering from thrombosis, i.e. thrombosis may be occurring in this stage of DIC. This stage is very difficult to diagnose because we lack suitable assays for detecting hypercoagulability (most screening assays of hemostasis are normal) and microvascular thrombi are difficult to detect clinically. The tests that may be abnormal are markers of thrombin generation, of which only D-dimer is readily available in animals. Over time (and depending on the nature of the initiating disease), spatial and temporal control over hemostasis is lost as inhibitors are overwhelmed, downregulated or inhibited, the endothelium becomes dysfunctional or injured, inflammation is stimulated and tissues are injured, with release of damage-associated molecular patterns (DAMPs), including extracellular or cell-free DNA, histones and nucleosomes (DNA-histone complexes), neutrophil proteases (elastase, myeloperoxidase) and other molecules (such as α-defensins, which act a DAMPs) and inflammatory nuclear-based cytokines (such as high mobility group box protein 1 or HMGB1). These DAMPs can activate coagulation (e.g. cell-free DNA), injure other tissues and upregulate TF (e.g. HMGB1). Thrombin generation then proceeds unchecked and is in the maintenance or perpetuation phase. DIC has now become “uncontrolled” and is in the “overt” stage, where thrombosis is occurring and platelets and coagulation factors are being consumed (but not excessively enough to always result in hemorrhage). During this stage, microvascular thrombi are forming, and platelets and coagulation factors are being consumed in clot formation. The animal is still hypercoagulable (prothrombotic) and is now in the thrombotic phase of “overt” DIC, particularly if fibrinolysis is inhibited. Again, this phase is challenging to diagnose, but serial testing of hemostasis may reveal worsening (decreasing activity of inhibitors, progressive increase in clotting times, decreasing platelet count, increasing D-dimer) or abnormal test results (various combinations of thrombocytopenia, prolonged APTT, prolonged PT, decreased antithrombin [AT] activity). Eventually, platelets and coagulation factors become depleted and fibrinolysis may dominate, such that the animal enters the hypocoagulable phase or hemorrhagic phenotype of DIC, in which clots do not form sufficiently, so bleeding dominates the clinical picture and is readily detectable (this occurs particularly in dogs). At this stage of DIC, laboratory tests will be abnormal, facilitating the diagnosis of DIC.
Note, that DIC fuels or perpetuates its own fire…inflammation is one of the main inciting causes of DIC and activated coagulation factors can induce an inflammatory response, including via activation of complement. Similarly, inhibitors have anti-inflammatory reactions so the loss of inhibitors and generation of activated coagulation factors and activation of platelets all serve to exacerbate the inflammatory state, which then, in turn, accelerates and promotes activation of hemostasis. This creates a vicious cycle and helps convert DIC from the “non-overt” into an “overt” phase. Note that non-overt DIC does not always naturally segue into overt DIC. For instance, massive head trauma may immediately result in overt DIC from release of tissue factor in the brain (although some categorize this condition separately from DIC), whereas low-grade inflammation may incite a more slow-burning controlled process (non-overt DIC) that may not progress.
Although hemorrhage is the most apparent clinical manifestation of DIC, microthrombi have far more serious sequelae, due to the effects of hypoxic injury on end-organ function, which exacerbates DIC. It is usually end-organ injury or failure that results in the demise of the patient and DIC should be thought of first as a thrombotic syndrome and second as a hemorrhagic one. Which phase of DIC dominates depends on the initiating cause and species. Dogs have robust fibrinolysis so the hemorrhagic phenotype of DIC dominates. In contrast, hemorrhage is uncommon in cats or horses with this disorder, suggesting suffer more from the thrombotic phenotype of DIC. Indeed, the disorder has been called “sub-clinical” in these species (which is really a misnomer, because thrombi are having clinical consequences, just ones that are hard to definitively ascribe to thrombosis, e.g. ischemic injury will contribute or cause azotemia, dyspnea, hepatic injury, etc).
An alternative way to think of DIC is via clinical manifestations. Some authors have proposed DIC be described as a bleeding or hyperfibrinolytic phenotype, which typifies the coagulopathy of acute trauma (now being considered its own entity to some degree [Gando et al 2013]) and obstetrical complications (e.g. amniotic rupture) in humans; a thrombotic or hypercoagulable phenotype (e.g. bacterial sepsis in humans); and a bleeding phenotype, which consists of both hypercoagulability with concurrent bleeding secondary to consumption versus hyperfibrinolysis alone (Wada et al 2014). The key concept of this phenotype-driven DIC is that the underlying disorder triggers particular secondary hemostastic disorders, which are governed by the balance between fibrinolysis and hypercoagulability and thrombosis.
Phases of DIC
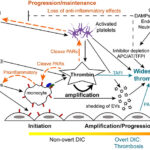
Over the past decade, our understanding of DIC has evolved from recognition of a severe hemorrhagic disorder to appreciation of DIC as a disease continuum. The process is initiated by inappropriate activation of hemostasis, at first kept in check by natural inhibitors. If regulatory mechanisms become overwhelmed, DIC progresses to a full-blown decompensated disorder characterized by systemic thrombosis and ultimately, particularly in dogs, a consumptive coagulopathy, the most readily recognized form of DIC in the clinical situation. For ease of understanding, the complex process that is DIC, can be broken down into the phases of initiation, amplification and progression/maintenance/perpetuation. Although there is always an initiating phase (which may be continuously active, even at a low level), the amplification and progression phases can proceed simultaneously depending on the underlying cause. DIC is all about thrombin generation, which can be divided into these stages: Initiation, amplification, progression, and dissemination
Initiation of thrombin generation
An important aspect of DIC is that it is never a primary disease. It is always initiated or triggered by another disease process. Diseases that can induce DIC are given in the table below, along with potential mechanisms by which those diseases can trigger DIC. The most common diseases associated with DIC are moderate to severe inflammation, neoplasia, bacterial sepsis, and massive endothelial injury. DIC has been documented in most domestic animals, with the exception of camelids. In dogs and cats, neoplasia and systemic inflammation (e.g. sepsis, pancreatitis, IMHA, heat stroke) are the most common initiating diseases. Endotoxemia (secondary to strangulating or inflammatory gastrointestinal disorders) and sepsis are the main causes of DIC in adult horses and neonatal foals, respectively. Similarly, DIC is primarily due to endotoxemia or sepsis in ruminants (such as secondary to mastitis or metritis).
DIC is thought to be initiated in these diseases primarily by the excessive exposure of extravascular tissue factor (TF), through massive endothelial injury or aberrant expression of TF on cells within the vasculature, e.g. monocytes, endothelial cells, cancer cells. Inflammatory cytokines have been shown to upregulate TF on monocytes and likely endothelial cells. Decryption of hidden forms of TF versus increased synthesis contributes to upregulated expression on cell surfaces and shed EVs. Decryption is thought to involve formation of phosphatidylserine-rich microdomains in membrane surfaces. Cancer cells, and their shed EVs (including exosomes and membrane-derived microvesicles), can constitutively express tissue TF. There are also TF-independent ways by which hemostasis could be activated in conditions associated with DIC. This includes the expression of proteases, such as a factor X-activating enzyme, on cancer cells.
New evidence also suggests that factor XII and kallikrein (of the so-called “contact: pathway) can promote activation of FIX and prothrombin, in a FXI-independent manner, in the presence of negatively charged long-chain polyphosphates, such as those found in bacteria (Puy et al 2013). In addition, activated neutrophils and other cell types (e.g. cancer cells, dying cells) can release DAMPs within nuclear material (extracellular or cell-free DNA, histones) and mitochondria (cell-free DNA), which is procoagulant and antifibrinolytic. For instance, cell-free DNA, by virtue of its negative charge, can activate FXII of the intrinsic pathway. Histones can directly activate prothrombin to thrombin (albeit slow), activate FXI in the presence of platelets, and also activate platelets (Gould et al 2015). This could be an additional mechanism of DIC initiation in disorders, such as bacterial sepsis, severe acute inflammation and cancer (Gould et al 2015, Liaw et al 2015).
| Disease | Potential mechanism of DIC initiation |
| Infectious agents: Bacterial sepsis (gram positive and negative bacteria), viruses (e.g. Feline infectious peritonitis), protozoa (Babesia canis), parasites (e.g. Angiostrongylus vasorum), rickettsia (e.g. Rocky Mountain Spotted Fever) | TF expression: Pro-inflammatory cytokines and lipopolysaccharides in gram-negative bacteria induce TF expression on monocytes and (likely) endothelial cells. Small amounts of TF can be expressed in platelets and other cells, such as eosinophils, however monocyte-derived TF is thought to be the main mechanism of DIC initiation in bacterial sepsis. Damage-associated molecular patterns (DAMPs) within nuclear material released by activated neutrophils, injured or dying cells can also promote endothelial and monocyte TF expression (e.g. the nuclear pro-inflammatory cytokine, HMGB1) and shedding of TF-expressing extracellular vesicles (directly or through inflammatory cytokine production). Tissue factor can also be expressed and shed from dying cells. Downregulation of inhibitors: Inflammatory cytokines can downregulate the production of inhibitors directly, such as thrombomodulin, antithrombin, protein C, or affect their activation (e.g. altering heparin like glycosaminoglycans in the endothelial glycocalyx which affects antithrombin and tissue factor pathway inhibitor [TFPI] activity). Activation of factor XII (FXII; contact pathway) or FXI (intrinsic pathway): In the presence of long chain bacterial polyphosphates, FXII can activate factor IX and prothrombin directly (Puy et al 2013) and also inhibit fibrinolysis (all thrombus-promoting activities). Extracellular or cell free DNA (but not necessarily that incorporated into nucleosomes or NETs [Noubouossie et al 2019]) or nuclear material can also activate FXII and initiate coagulation via the intrinsic pathway. This DNA material is released in abundance by neutrophils in response to bacteria and activated platelets, in a process called NETosis (Liaw et al 2015). Histones can also activate FXI without FXII in a platelet- and platelet short chain polyphosphate-dependent manner and can also convert prothrombin to thrombin (but this is rather slow and unlikely to be of much relevance in vivo) (For more on this, see review on NETs and coagulation by Alhamdi and Toh 2017 and Noubouossie et al 2019). Endothelial injury: Exposure of extravascular TF on fibroblasts and smooth muscle cells, induction of TF on endothelial cells, and release of nuclear material can cause a vicious cycle by causing endothelial cytotoxicity. Nuclear material (particularly histones) and bradykinin (generated by FXII and prekallikrein activity) can also induce vascular permeability, leading to a loss of inhibitors into the extravascular space. Histones can stimulate release of ultra-high molecular weight von Willebrand factor “strings” from Weibel-Palade bodies in endothelial cells, which then capture and potentially activate platelets, helping promote thrombus formation and contributing to thrombocytopenia in sepsis (Michels et al 2016). Platelet activation: Bacteria can activate platelets via toll-like receptor 4, which can stimulate thrombin generation (Matus et al 2017). Thrombin generation could also be mediated by platelet-associated TF expression or release of platelet polyphosphates (which can directly activate FXII). Tissue injury from bacterial infection releases DAMPs, which also activate platelets. Platelet activation can contribute to thrombocytopenia in sepsis, including formation of platelet-derived microthrombi. Complement activation: Lectins in pathogens are recognized by mannin binding lectin, which generates MBL-associated serine proteases or MASPs, which activate complement via the lectin pathway (produces the C3 convertase via C4 cleavage). Pathogens can also be recognized by C1q in the classic pathway, which also forms the C3 convertase via cleavage of C4. MASPs can act as coagulation factors, generating thrombin from prothrombin and forming fibrin from fibrinogen. The membrane attack complex and anaphylatoxin, C5a, upregulate TF expression on monocytes and endothelial cells and activate platelets. |
| Cancer: Particularly metastatic tumors, but also hemangiosarcoma and hematopoietic (lymphoma, acute leukemia) neoplasia | Expression of TF on cancer cells: Constitutive or induced by hypoxia, cytokines, or apoptosis. Induction of TF expression: Tumor- or host-secreted cytokines act on monocytes, fibroblasts, and endothelial cells. Shedding of procoagulant membrane-derived EVs: TF- and phosphatidylserine (PS)-bearing microparticles from tumor cells, hematopoietic cells, other cell types. Expression/secretion of procoagulants: Cancer procoagulant (a vitamin K-dependent cysteine protease) and mucin can activate coagulation factors, released polyphosphate and extracellular nuclear material can activate FXII (Nickel et al 2015). Chemotherapy-induced changes: Tumor lysis (upregulation of TF, expression of PS, shedding of EVs, release of procoagulant factors, such as cell-free DNA), may be exacerbated by myelotoxicity (thrombocytopenia). |
| Inflammation/necrosis: Trauma, immune-mediated hemolytic anemia (IMHA; dogs), pancreatitis, heat stroke, hepatitis, vasculitis, gastric dilatation-volvulus (dogs), strangulating obstructions and inflammatory gastrointestinal disorders (horses) | NETosis: Suicidal or non-suicidal release of nuclear DAMPs by activated neutrophils can activate hemostasis through FXII and FXI (see above). They also activate platelets. Histones and cell-free DNA can also downregulate or decrease the activity of anticoagulants (e.g. decreasing of thrombomodulin, direct binding and inhibition of Protein C) and cause release of TF- and PS-expressing procoagulant EVs (see Alhamdi and Toh review). Extracellular DNA and histones can also affect fibrinolysis, usually inhibiting it (promoting thrombosis), and confer resistance to fibrinolysis by making a stronger clot (Yong et al 2023). Neutrophil enzymes bound to the released DNA can cleave coagulation factors, fibrin and inhibitors. α-defensins are also antifibrinolytic and promote formation of a stronger clot (Abu-Fanne et al 2019). Bacterial polyphosphate and polyphosphate release by platelets (shorter chain than those seen in bacteria) can contribute locally to thrombosis and inhibition of fibrinolysis. Histones can also directly activate prothrombin to thrombin as an “alternate”, albeit less efficient, prothrombinase, which is both phospholipid- and FVa-independent (Yong et al 2023). Note that neutrophils are not the only cells that can undergo NETosis – other cell types can as well, including eosinophils and monocytes and cancer cells and injured cells can also release DAMPs. Inflammatory cytokines: See infectious agents above. These also induce NETosis and NETs can stimulate pro-inflammatory cytokine release. Endothelial injury: See infectious agents above. Tissue injury/necrosis/apoptosis: Exposure of TF; activation of FXII via extracellular DNA and histones, shedding of PS-enriched EVs by apoptotic cells or induced by histones. Enzymes released from tissues (e.g. trypsin from the pancreas in pancreatitis) can also activate and cleave factors, including inhibitors and fibrin. Release of procoagulants: See above. Enzyme release: Elastase released from neutrophils binds to NETs during NETosis and also cleave coagulation factors, cross-linked fibrin (releasing D-dimer) and inhibitors (antithrombin, tissue factor pathway inhibitor). Complement activation: DAMPs released by tissue injury can activate the classical (C1q) and lectin pathways of complement, which can facilitate activation of coagulation and platelets. See above. |
| Intravascular hemolysis: IMHA, acute transfusion reactions, insect/snake bites, oxidant injury (e.g. red maple leaf toxicity in horses | Endothelial injury: See mechanism above Erythrocyte procoagulant activity: RBC membrane-associated elastase, eryptosis with expression of PS and shedding of PS-enriched EVs, formation of meizothrombin Complement activation: Antibodies can activate complement. See above. |
| Envenomation | Snake venom proteases: Direct activation of coagulation factors (e.g. factor X) and cleavage of fibrinogen, phospholipase-induced tissue injury/cell lysis. |
Amplification of thrombin generation
Amplification of thrombin generation occurs through thrombin activating cofactors and intrinsic pathway factors and occurs on the surface of activated cells, such as platelets and leukocytes, and their shed EVs. DNA scaffolds can also serve as a procoagulant surface that supports thrombin amplification. Thrombin amplifies its own production by activating the following:
- Factor XI of the intrinsic pathway of coagulation. This then cleaves and activates factor IX. Release of polyphosphates from activated platelets promote the activation of FXI by thrombin.
- Factor VIII: The intrinsic pathway cofactor for activated FIX (part of the intrinsic “tenase” complex). Activated FVIII markedly accelerates activation of factor X on the surface of PS-expressing cells.
- Factor V: The common pathway cofactor for activated FX (part of the “prothrombinase” complex). Like FVIIIa, FVa markedly accelerates thrombin generation by FXa on the surface of cells, producing bursts of thrombin, that can cleave fibrinogen to fibrin. Polyphosphates promote factor V activation (Smith et al 2006).
- Platelets: Thrombin binds to protease-activated receptors (PAR) on platelets. Activated platelets recruit additional platelets, release short-chain polyphosphates and other agonists (e.g. ADP), and flip their membranes, expressing PS on their outer membrane leaflet, thus providing a scaffold and localizing area for coagulation factor assembly. PS also positions coagulation factors in an optimal conformation so coagulation factor activation is accelerated (along with thrombin generation). Activated platelets also shed PS-expressing EVs, which are strongly procoagulant (more than the platelet itself) and vastly increase the surface area on which thrombin generation proceeds. Although platelets are considered the main source of PS and surface membranes that support thrombin generation, activated leukocytes and RBC also provide PS-bearing membranes and shed EVs and likely play a major role in DIC. For instance, histones have been shown to cause PS expression on RBCs, turning them into procoagulant molecules, and are also platelet agonists (Gould et al 2015). Conversely, activated platelets can also stimulate NETosis (see Alhamdi and Toh review).
- DNA/histones: Not only do these activate FXII, but they can also serve as a negatively charged surface on which coagulation proceeds (see Alhamdi and Toh review).
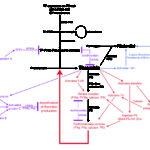
Thrombin not only amplifies its own generation, but it also creates a fibrin clot, by first cleaving fibrinogen to form soluble fibrin and then activating factor XIII, which crosslinks soluble fibrin to insoluble fibrin. At the same time that thrombin is promoting clot formation, it is also inhibiting clot breakdown, by activating an inhibitor of fibrinolysis (thrombin-activatable fibrinolysis inhibitor or TAFI). The activity of TFPI is enhanced by polyphosphates, which also increase fibrin clot strength making the fibrin clot more resistant to lysis (Smith and Morrissey 2008). In early DIC, as in physiologic hemostasis, thrombin’s generation and its actions are opposed by the plasma anticoagulants, antithrombin (AT) and protein C, and the endothelial cell surface receptors, thrombomodulin (which activates protein C) and endothelial protein C receptor (which modulates protein C activation and also triggers downstream endothelial signaling that can be cytoprotective). This phase of systemically activated coagulation restrained by natural anticoagulants is referred to as “non-overt” DIC, a state of stressed, but compensated hemostasis. As indicated previously, patients with non-overt DIC are hypercoagulable, i.e. at risk for widespread microvascular fibrin deposition.
Progression/maintenance of thrombin generation
The development of DIC depends not only on the location and severity of the inciting stimulus, but on the ability of naturally occurring inhibitors to regulate the procoagulant hemostatic response. These inhibitors hold the breaks on hemostasis. Effective inhibition or containment results in the non-overt or compensated stage of DIC and DIC may never progress beyond that point. However, depending on the primary disease, severity of the process and extent of hemostasis activation, DIC can progress from the non-overt to overt phase or DIC can be maintained despite treatment of the primary disorder. The evolution of DIC from a non-overt to overt phase, progression during non-overt or overt phases, and persistence of DIC after treatment of the initiating disorders is likely mediated through two-way potentiating interactions between inflammation and hemostasis and endothelial dysfunction, which also contributes to dissemination.
Overt DIC is characterized by:
- Loss of control or inhibition: Inhibitors become overwhelmed, are inactivated or are consumed in trying to stop the excessively activated hemostatic system (see table below). The unchecked and systemic generation of thrombin produces diffuse microvascular fibrin thrombi, the hallmark of the hypercoagulable phase of overt DIC.
- Tissue factor pathway inhibitor (TFPI): Under physiologic hemostasis, the TF-FVIIa-FXa complex is rapidly neutralized by TFPI. TFPI is primarily expressed on endothelial cells, with its activity enhanced by cell-surface heparin and heparin-like glycosaminoglycans. In the process of DIC, TFPI is rendered ineffective through a number of mechanisms, including cleavage of TFPI by granulocytic elastases (whose activity are promoted by cell free DNA), cytokine-mediated suppression of TFPI expression, decreased protein S (a cofactor for TFPI, see below), and generation of excess TF-FVIIa that overwhelms TFPI’s inhibitory capacity.
- Antithrombin: Pro-inflammatory cytokines downregulate production in hepatocytes (this does not appear to occur in cats). Antithrombin is also consumed by binding to thrombin, forming thrombin-antithrombin (TAT) complexes. Increased numbers of TAT complexes are proof of excessive production of thrombin and a hypercoagulable state. In one study, the median concentration of TAT complexes increased with the number of abnormal hemostasis test results (thrombocytopenia, prolonged PT or APTT, hypofibrinogenemia, increased FDPs, and low AT activity) in 247 dogs with various diseases. Higher median TAT complexes were seen in dogs with 1 or more abnormal hemostasis test results, including thrombocytopenia (<200,000/uL), hypofibrinogenemia (<178 mg/dL), PT and/or APTT >25% prolonged, high FDP (>10 ug/mL) and low AT activity (<90%). Median values were significantly higher in dogs with 2 or more abnormalities (Rimpo et al 2018). Downregulation of heparin-like glycosaminoglycans on endothelial cells also decrease the activity of antithrombin. As for TFPI, neutrophil-derived and other proteases can degrade antithrombin.
- Protein C: Protein C is produced in the liver and its production is downregulated by proinflammatory cytokines. Protein C is activated by thrombin binding to thrombomodulin, with the activity being promoted by the endothelial protein C receptor. Inflammatory cytokines also decrease endothelial expression of thrombomodulin and the endothelial protein C receptor, decreasing activation of protein C in DIC. Histones released from the nucleus of cells (cancer cells, dying cells, neutrophils) can bind to both thrombomodulin and protein C, decreasing protein C activation (Gould et al 2015). Decreased levels of protein S, a co-factor for protein C, (see below), also contribute to decreased activity. Neutrophil proteases (and other proteases, such as trypsin or plasmin) can potentially degrade protein C. A protein C inhibitor binds to activated protein C and facilitates its clearance – this clearance is increased in DIC.
- Protein S: This vitamin K-dependent protein produced in the liver is a co-factor for protein C and TFPI. It is found in free and complexed form to C4-binding protein. C4-binding protein is a positive acute phase reactant and increased levels in inflammation decrease the amounts of free protein S that can behave as a cofactor for other inhibitors. As for other inhibitors, proteases can degrade protein S.
- Tissue factor pathway inhibitor (TFPI): Under physiologic hemostasis, the TF-FVIIa-FXa complex is rapidly neutralized by TFPI. TFPI is primarily expressed on endothelial cells, with its activity enhanced by cell-surface heparin and heparin-like glycosaminoglycans. In the process of DIC, TFPI is rendered ineffective through a number of mechanisms, including cleavage of TFPI by granulocytic elastases (whose activity are promoted by cell free DNA), cytokine-mediated suppression of TFPI expression, decreased protein S (a cofactor for TFPI, see below), and generation of excess TF-FVIIa that overwhelms TFPI’s inhibitory capacity.
- Dissemination: This is where there is loss of spatial localization of hemostasis. Instead of being restricted to a specific site of injury, it occurs throughout the vasculature. This can be triggered by massive endothelial injury, intravascular expression of TF but can also be facilitated by the release of procoagulant EVs and endothelial and tissue damage induced by microthrombosis, DAMPs, and vasoactive mediators (e.g. bradykinin, endothelin). Procoagulant EVs provide an expanded surface on which coagulation proceeds. Although they have a short half life of a few minutes, circulating TF-expressing EVs are incorporated early into developing thrombi and promote thrombosis. In addition, tumor-derived EVs can induce thrombosis without vascular injury (see review by Owens and Mackman 2011). During physiologic and pathologic hemostasis, activated platelets are thought to be the main sources of membrane-surface PS and shed EVs in circulation, however leukocytes, injured cells, cancer cells and RBCs are also likely sources of procoagulant EVs, particularly in DIC (see reviews by Owens and Mackman 2011, Reid and Webster 2012). These EVs also act intercellular messengers and it is possible that, through fusion and transfer of their intracellular cargo and induction of oxidant and inflammatory responses, EVs contribute to widespread endothelial and organ injury in DIC (Reid and Webster), which helps DIC progress or be maintained/perpetuated. Similarly, histones can induce distant endothelial and tissue injury from their site of origin. Developing or formed fibrin thrombi or microvascular platelet plugs will also contribute to endothelial and tissue injury.
- Perpetuation of an activated coagulation system:
- Inflammation and hemostasis cross-talk: A self-perpetuating cycle develops in DIC triggered by sepsis or non-septic inflammation. Inflammation initiates coagulation via tissue injury and release of DAMPs, inflammatory cytokine-induced TF expression, complement activation, and platelet and leukocyte activation and microvesiculation. Coagulation, in turn, incites inflammation, through the proinflammatory and complement-activating properties of active coagulation factors, activated platelets and endproducts of thrombin and fibrinolytic activity (e.g. fibrin and its degradation products). For instance, thrombin binds to and activates protease-activated receptors (PARs) on platelets, monocytes and ECs, stimulating release of proinflammatory mediators and inducing adhesion molecule expression. Thrombin-mediated cleavage of PARs can lead to endothelial dysfunction and vascular leakage and increase exposure of subendothelial TF, powering coagulation. Activated platelets also contribute to the crosstalk between inflammation and coagulation. They release inflammatory mediators, such as platelet-activating factor and HMGB1, activate and bind to complement, bind to and recruit leukocytes and stimulate NETosis (see reviews by Gando et al 2016 and Iba and Levy 2018). Several coagulation inhibitors, including protein C and AT and the endothelial receptors, thrombomodulin and endothelial protein C receptor, have anti-inflammatory properties and endothelial cytoprotective properties, which may be lost in DIC. The release of vasoactive peptides (e.g. bradykinin) from damaged or cytokine- or thrombin-stimulated cells induces hemodynamic changes that often decrease tissue perfusion, thereby contributing to hypoxic injury. Secondary acidosis inhibits platelet function and coagulation factor activity, furthering development of hemorrhagic DIC.
- Endothelial dysfunction: This causes a loss of inhibition and a gain of proinflammatory and procoagulant properties. Healthy endothelial cells secrete potent platelet inhibitors (e.g. prostacyclin, ADPase) and express receptors, including PARs, thrombomodulin and endothelial protein C receptor, that modulate coagulation and inflammation. As a direct consequence of the primary disease or as a sequela to DIC, a variety of factors, including pro-inflammatory cytokines (e.g. HMGB1), ischemia, histones, EVs, and acidosis, cause loss of the normal protective functions of endothelial cells. Affected endothelial cells may become proinflammatory (secrete inflammatory cytokines and upregulate adhesion molecules) and procoagulant (express TF, release platelet activating compounds) with decreased anticoagulant activity (e.g. cytokine-mediated downregulation of thrombomodulin, degraded glycocalyx with reduced expression of glycosaminoglycans).
- Other tissue or organ injury: Injury to and dysfunction of organs other than the endothelium can also contribute to perpetuation of DIC, through DAMP release (histones), induced TF and microvesiculation of necrotic or apoptotic cells, and stimulation of inflammation.
| Mechanism | Causes |
| Decreased hepatic synthesis of AT, protein C, protein S | Hepatic injury (e.g. ischemia, histones), pro-inflammatory cytokine-mediated downregulation |
| Decreased activity of TFPI, AT, protein C, protein S | Low protein S: This is from increased C4-binding protein, a positive acute phase reactant protein which binds protein S. Lack of free protein S decreases protein C and TFPI activity. Polyphosphates: Inhibit TFPI. Decreased protein C activation: Due to downregulated thrombomodulin and endothelial protein C receptor by pro-inflammatory cytokines (e.g. HMGB1), DNA and histones. Thrombomodulin and endothelial protein C receptor can also be cleaved by neutrophil elastases. Endothelial dysfunction: Decreased AT and TFPI activity (altered glycocalyx, e.g. down-regulation of heparin-like glycosaminoglycans by inflammatory cytokines and histones). |
| Increased consumption of AT, protein C | Rapid clearance of thrombin-antithrombin complexes and activated protein C-protein C inhibitor complexes. |
| Loss of TFPI, AT, protein C, protein S | Vascular leakage due to release or production of vasoactive mediators (e.g. bradykinin, histones). |
| Degradation of TFPI, AT, protein C, protein S | Proteases, e.g. neutrophil elastases, bacterial peptides (e.g. the Pla omptin), histones (degrade TFPI). |
Fibrinolysis
Although not considered part of the phases of DIC, the balance between antifibrinolytic and profibrinolytic forces may actually dictate outcome in terms of clinical manifestation of DIC as thrombosis or hemorrhage. Multiple factors affect fibrinolysis in DIC, shifting the balance from a fibrinolytic and more hemorrhagic phenotype to an antifibrinolytic or more thrombotic phenotype and vice versa.
- Profibrinolytic: Shifting of the balance towards profibrinolytic forces will lead to excessive breakdown of clots and hemorrhage. This could be secondary to increased tPA or urokinase-type plasminogen activator (uPA, normally operates extravascularly but can contribute to fibrinolysis in DIC) release from endothelial cells or cancer cells (for uPA). In sepsis, this profibrinolytic response occurs early but is rapidly swamped by the antifibrinolytic actions of PAI-1. In addition, activation of FXII will stimulate fibrinolysis because both FXII and kallikrein can act as weak plasminogen activators and they cleave high-molecular weight kininogen to bradykinin. Along with being a potent vasodilator, bradykinin is also one of the most potent stimulators of tPA release. Histones may also stimulate tPA release and this may contribute to the hyperfibrinolytic phenotype of massive bleeding characteristic of trauma-induced coagulopathy. There is controversy as to whether the latter is a variant of DIC (Hayakawa 2017) or its own unique entity (Cohen and Christie 2017). Inhibition or decreased fibrinolytic inhibitors, such as thrombin-activatable fibrinolysis inhibitor (TAFI), PAI-1 and antiplasmin, would also promote fibrinolysis. There are also species differences with fibrinolysis, in that dogs naturally have higher tPA activity than humans (Lanevschi er al 1996). This may also explain the propensity for dogs to present with hemorrhage in DIC (although thrombosis is still likely present) versus other species. Activated protein C is also profibrinolytic, in a calcium, phospholipid and Protein S-dependent manner (de Fouw et al 1990).
- Anti-fibrinolytic: In sepsis, antifibrinolytic mechanisms, mostly through injury-stimulated release of PAI-1 from endothelial cells, dominate in murine models and humans, which is why DIC mostly manifests as thrombosis in sepsis (Gando et al 2016). Horses also appear to be similar to humans in that increased concentrations of PAI-1 are found in colic and correlate with endotoxemia (Collatos et al 1994). Nuclear material (cell free DNA, histones and NETs) and polyphosphates (from bacteria or platelets) and α-defensins from neutrophils are also anti-fibrinolytic. These compounds also promote the formation of a stronger fibrin clot, which is more resistant to fibrinolysis. Nuclear material also inhibits tPA-mediated conversion of plasminogen to plasmin and polyphosphates act as cofactors for thrombin-mediated activation of TAFI (Alhamdi and Toh 2017).
Diagnosis of DIC
Every animal with an inciting primary disease should be considered at risk for developing DIC. Confirmation of DIC is challenging because animals may be examined at any point of the DIC continuum, clinical signs may be subtle or nonspecific, and individual laboratory tests are variably sensitive and none are specific for this syndrome. Rather, a constellation of abnormal test results are used to identify DIC and help discriminate between overt and non-overt forms. Histopathologic evidence of fibrin thrombi in the microvasculature in biopsy or necropsy specimens can be definitive, however only small sections of tissues are evaluated for thrombi and .fibrin thrombi often lyse rapidly after death. Thrombi can be detected with phosphotungstic acid hematoxylin and immunohistochemical staining, however the two techniques vary in their sensitivity (Cotovio et al 2007, Cesarini et al 2016). Ischemic necrosis can be a surrogate for fibrin thrombi, but is not specific for DIC, being seen with ischemia due to other causes, including vessel torsion, rupture, hemodynamic changes and anemia. Thus, histopathologic examination of tissues cannot be relied upon to diagnose or confirm DIC, especially antemortem.
The table below summarizes the differences between non-overt and overt stages of DIC, which can be discriminated, to some extent on laboratory findings and also clinical signs.
Scoring criteria for the diagnosis of overt and non-overt DIC have been developed by the ISTH and British, Italian and Japanese Hemostasis and Thrombosis or Hematology societies (Taylor et al 2001, Wada et al 2013). These schemes have allowed for a degree of standardization across studies and, in 2013, the Scientific and Standardization Committee on DIC of the ISTH recommended the use of grading schemes, albeit different, for diagnosis and treatment of DIC. The Committee on DIC also recommended the use of serial testing (based on moderate quality of evidence) (Wada et al 2013). Only 2 studies have evaluated scoring criteria for the diagnosis of overt DIC in dogs and both showed an association between DIC diagnosis and mortality.(Wiinberg et al 2010, Goggs et al 2018) Since the diagnosis of DIC still varies between published studies in animals, there is a still need for an evidence-based approach to standardize criteria for the diagnosis of DIC, which may differ between species.
Non-overt DIC
As described above, non-overt DIC is characterized by an activated, but not overwhelmed, hemostatic system. Non-overt DIC also applies to the designation of hypercoagulability (which can be thought of being “at risk” of thrombosis) or “chronic DIC” reflecting continuous low grade, but compensated, activation of coagulation (see table below).
- Clinical signs: Affected animals should have an underlying disease predisposing them to DIC. Since this phase is dominated by thrombosis (if occurring at all), it is difficult to impossible to detect clinically, particularly when thrombi occur internally or in small vessels (the majority of thrombi with DIC). Doppler ultrasonography may be useful in detecting larger thrombi. Blood gas analysis can detect ventilation:perfusion mismatch in patients with dyspnea and support pulmonary thromboembolism. Deteriorating organ function, e.g. worsening azotemia, increasing liver leakage enzymes, hyperlactatemia, could also be due to hypoxic injury to major organs.
- Laboratory tests: Routine coagulation screening tests, such as the APTT and PT, are insensitive to this phase because they detect coagulation factor deficiencies, rather than accelerated coagulation or hypercoagulability. Non-overt DIC is marked by excessive thrombin generation and tests geared towards detection of excessive thrombin are useful for detection of this phase. This might include measurement of TAT complexes (Rimpo et al 2018), D-dimer, and sophisticated assays that measure the kinetics and rate of thrombin generation, e.g. thrombin potential assays (Cuq et al 2018). Unfortunately, with the exception of D-dimer (which is not specific for DIC), these tests are not widely available for routine clinical diagnosis. It was hoped that viscoelastographic or elastometric techniques, which monitor and record the rate of fibrin formation in whole blood, would be useful for detecting this phase of DIC, but they are yet to demonstrate this promise and are, unfortunately affected by other variables (including sample collection and method of activation [deLaforcade et al 2014, Flatland et al 2014, Hanel et al 2014], which are frequently present in dogs with DIC (e.g. anemia -e.g. hypercoagulable tracings are present in dogs and horses with lower hematocrits) (McMichael et al 2011, Smith et al 2012, McMichael et al 2014). There are several human schemes to help diagnosis of non-overt DIC. All rely on documentation of an underlying disease and various combinations of routine test results (Taylor et al 2001, Wada et al 2013) The most optimal technique may be serial measurement of coagulation assays to document a worsening, stable or improving hemostatic system. Worsening of hemostatic dysfunction (progressive prolongation of APTT, decreasing platelet count) even in the absence of test abnormalities (APTT and platelet count may still be within reference intervals) would support non-overt DIC and potential progression to overt DIC in a patient with an associated primary disease process.
Overt DIC
Overt DIC means that the disorder has manifested clinically, i.e. thrombosis is definitely occurring and some animals may be suffering from hemorrhage. Widespread microvascular thrombosis decreases blood flow to vital tissues causing hypoxic injury, cell death, and organ failure, which can be responsible for the high morbidity and mortality of DIC, particularly in horses. Although severe signs at presentation often prompt laboratory investigation, screening tests to detect overt DIC should be performed in all patients with primary inciting disorders. There is no single diagnostic test for overt DIC, rather we rely on a combination of test abnormalities, along with associated clinical signs (if present) and documentation of a primary disease. A retrospective study of 804 ill dogs in which coagulation testing was performed showed that criteria for diagnosis of DIC of at least 3 abnormal test results (low platelet count, prolonged PT, prolonged APTT, low AT activity, low fibrinogen concentration or high D-dimer concentration) based on institution-derived reference intervals was the most sensitive method for determining outcome (survivor versus non-survivor) (Goggs et al 2018).
- Clinical signs: Affected animals should have an underlying disease predisposing them to DIC. Depending on the species, this phase can be dominated by thrombosis (thrombotic phenotype), which is difficult to detect, or hemorrhage, which is far readily apparent clinically. Hemorrhage has typically been attributed to a consumptive coagulopathy with coagulation factor deficiencies, but there is a recent paradigm shift, in that hemorrhage is now thought to be due to endothelial dysfunction with gaps developing in intact endothelium (e.g. from inflammatory cytokines and vasoactive mediators). Platelet dysfunction is thought to contribute to the hemorrhage (platelets are required for vascular integrity and one platelet can plug minute endothelial gaps) (Puetz 2021, Thachil 2021). Hemorrhagic DIC develops frequently in dogs, but appears to be uncommon in horses and cats. This could be due to the robust fibrinolysis in dogs (they have high tissue plasminogen activator activity [Lanevschi et al 1994]) and perhaps, inhibition of fibrinolysis (by upregulation of plasminogen activator inhibitor, as occurs in humans) in other species, as documented in horses with colic (Collatos et al 1994); this would promote thrombosis. Regardless, in all species, fibrin thrombosis is occurring in this phase of DIC, so DIC should be thought of first and foremost as a thrombotic and not a hemorrhagic disorder. When bleeding occurs, it manifests as any (and more than one) of the following: epistaxis, petechiae, bruising, prolonged bleeding after venipuncture or minor surgical procedures, and spontaneous hematoma formation and body cavity hemorrhage.
- Laboratory tests: Diagnosis of overt DIC relies upon identifying abnormalities in multiple tests, rather than a single pathognomonic sign or laboratory finding. Traditional criteria for diagnosis of overt DIC in animals include a combination of 3 or more test abnormalities, specifically abnormalities in all pathways of hemostasis, showing activation of coagulation (thrombocytopenia, prolonged PT and/or APTT, hypofibrinogenemia), inhibitor consumption (low AT activity) and fibrinolysis (high D-dimer or FDPs). Note that RBC fragments are not included in these tests (although they are listed below, along with other non-routine tests), although they can be seen in some animals with DIC. Note that older studies used a combination of 2 or more abnormal findings (which included RBC fragments) for laboratory diagnosis of DIC, but the study by Goggs et al is the strongest evidence to date for the use of three or more abnormal test results (which is not to say that animals with fewer abnormal test results are not in DIC – overt or non-overt).
- Primary hemostasis: Mild to moderate thrombocytopenia is a consistent finding in dogs with overt DIC (75-100% sensitivity, i.e. some animals will not be thrombocytopenic [Bateman et al 1999, Stokol et al 2000]), but not in cats (personal observations) or horses (0-64% sensitivity [Dolente et al 2002, Stokol et al 2005]). Overt DIC is therefore unlikely in dogs having sequential platelet counts that remain stable and within reference intervals but should be suspected in dogs with a normal platelet count that is progressively declining (even while remaining within reference intervals) with serial measurement. In rare cases, some animals with overt DIC can have high platelet counts (if the underlying disease process, such as inflammation, is concurrently stimulating thrombopoiesis). Overt DIC is suspected in the latter animals if other assays on a DIC panel are abnormal (e.g. prolonged APTT, high D-dimer, low AT activity) and the animal has a disease process that could trigger DIC (of course). High concentrations of fibrin(ogen) degradation products may inhibit platelet function, so bleeding symptoms associated with primary hemostatic disorders (e.g. mucosal hemorrhage, petechiae) may occur even when the platelet count is not critically low (<10,000/uL).
- Secondary hemostasis: Prolonged coagulation times (PT, APTT, TCT) and hypofibrinogenemia may occur in DIC due to consumption of coagulation factors, including fibrinogen. Plasmin and other proteases (e.g. from neutrophils) may also cleave (inactivate) coagulation factors. The TCT may be additionally prolonged by high concentration of fibrin(ogen) degradation products which inhibit fibrin polymerization in the test (Mischke et al 2000). Remember, a low fibrinogen (<50 mg/dL in the dog and <90 mg/dL in other species; Marjory Brooks, personal communication) will prolong the PT and APTT, regardless of other coagulation factor deficits. The apparent diagnostic utility of these tests varies depending on species and stage at presentation. Of the routine coagulation screening tests, the APTT appears to be more sensitive for detecting DIC in animals than the PT or TCT (Bateman et al 1999, Estrin et al 2006, Stokol et al 2000 and 2005, Dolente et al 2002). Hypofibrinogenemia is generally an insensitive indicator of DIC because fibrinogen is an acute phase reactant protein and likely to be upregulated secondary to underlying inflammation (whether induced by DIC or the cause of DIC in the first place). Indeed, the finding of normal fibrinogen values in a patient with active inflammation suggests ongoing consumption of fibrinogen combined with increased in DIC, with the final concentration being determined by the balance between these two processes and which one is dominating. Fibrinogen concentration as measured by the Clauss clotting method (that used in the comparative coagulation laboratory) may also be underestimated in the presence of very high FDPs (Mischke et al 2000).
- Fibrinolysis: High fibrin(ogen) degradation product (FDPs) or D-dimer are characteristic of DIC in most species (Stokol et al 2000 and 2005). Even though fibrinolysis may be inhibited to some extent in DIC (e.g. due to increased plasminogen activator inhibitor-1 activity; Collatos et al 1994), it is still occurring, resulting in high values of these degradation products. In most diagnostic laboratories, D-dimer assays have replaced FDP testing for detecting fibrinolysis in DIC. D-dimer is a sensitive test for DIC in dogs (75-100%) but is less sensitive in horses (50%) (Stokol et al 2000 and 2005). D-dimer has not been adequately evaluated in cats, however internal studies show that D-dimer may only be increased in about 50% of cats with diseases predisposing them to DIC (pancreatitis, feline infectious peritonitis, cancer). In addition, D-dimer can be formed for reasons other than plasmin-mediated breakdown of crosslinked fibrin. For example, neutrophil proteases can also degrade crosslinked fibrin and the latter does not indicate excessive fibrinolysis per se (although with increased recognition of the role of NETs and neutrophils in coagulation, perhaps it truly does). In addition, gaps in endothelial cells causes leakage of large coagulation factors into the extravascular space and can result in extravascular fibrin production and lysis (potentially via urokinase plasminogen activator, which is usually tissue associated), leading to D-dimer formation that is not due to intravascular thrombosis (Thachil 2021).
- Decreased inhibitors: Low activity of inhibitors (AT and protein C) would be expected in overt DIC for the reasons given above. Low AT activity is one of the more sensitive tests for diagnosis of DIC in dogs (77-90%) and horses (89-93% (Stokol et al 2000 and 2005). In cats with DIC, AT activities may acutely decrease (Marjory Brooks, personal communication), however values rarely fall below reference intervals and may actually be high in some patients (Boudreaux et al 1989). So low AT activities should not be relied upon to diagnose DIC in cats.
- Red blood cell fragments: Additional tests sometimes used to characterize DIC include blood smear examination for erythrocyte fragments (schistocytes in particular but also keratocytes and acanthocytes – these are of most use in the dog, in which the combination of thrombocytopenia and red blood cell fragments should raise suspicion of DIC and prompt testing for this disorder). However, the absence of these fragments never rules out DIC as these RBC changes only occur in low numbers of animals (around 20% of cases in dogs [Feldman et al 1989] and in only 8% of cats [Estrin et al 2006). RBC fragments are very non-specific for DIC in cats, being seen more with various types of liver disease, including lipidosis (many of these cats are not in DIC). Similarly, fragments are not specific for DIC in dogs either and can occur with any other cause of blood turbulence causing shearing of RBC (e.g. portosystemic shunts, mitral valve disease), mechanical fragility of RBC (e.g. iron deficiency) and oxidant injury (keratocytes not schistocytes with oxidant injury) to RBC. RBC fragments are also rarely reported in horses and ruminants with DIC, although we have seen fragments in isolated cases of DIC in the latter species.
- Other tests: Other tests that have been performed in dogs are clot curve analysis (Richardson et al 2018) and thrombin-antithrombin complexes, which are increased in some dogs with 1 or more coagulation test abnormalities in one study using an in-house test for these complexes (which represent the most direct evidence of thrombin activation). In the latter study, the median TAT concentration increased with an increased number of coagulation abnormalities, supporting an association between thrombin generation and more severe coagulation defects (Rimpo et al 2018). However, none of these assays are common place in veterinary medicine. Thromboelastography/elastometry has also been performed in dogs with DIC and shows that both hypercoagulable and hypocoagulable tracings occur in dogs with DIC, with hypocoagulable tracings being associated with a higher risk of death (Wiinberg et al 2008, Barthélmy et al 2018). However, hypercoagulable tracings could be a direct consequence of the combination of anemia or high fibrinogen in affected dogs and hypocoagulable tracings will occur in thrombocytopenic animals, which is an inevitable consequence of DIC in dogs. Thus, it is unclear if thromboelastography provides any real additional information on hemostatic status in animals with DIC.
| Overt | Non-overt | |
| Alternate name | Uncompensated, acute, fulminant, consumptive coagulopathy, thrombotic phase = hypercoagulable, hemorrhagic phase – hypocoagulable | Compensated, chronic, subclinical, pre-DIC, hypercoagulable |
| Pathophysiology | Decompensated hemostasis with fibrin deposition throughout the microvasculature. Platelet and factor depletion develop over time. | Procoagulant excess opposed by coagulation inhibitors, partial compensation modulates fibrin deposition (thrombi may or may not be forming) |
| Clinical features | Primary inciting disease, thrombotic phenotype (systemic thrombosis, pulmonary thromboembolism, multiple organ dysfunction), hemorrhagic phenotype, such as excess bleeding from 1 or more sites (bleeding features of primary or secondary hemostasis) | Primary inciting disease, subclinical or tissue thrombosis (may not be clinically evident) |
| Screening test abnormalities | Thrombotic phase: Worsening test results or abnormal test results (see hemorrhagic phase). Hemorrhagic phase: Usually a combination of >2 of the following: Thrombocytopenia, prolonged clotting times (APTT, PT, TCT), hypofibrinogenemia, low AT, high FDP & D-dimer (tests vary in sensitivity and specificity depending on the species). May see RBC fragmentation (schistocytes, keratocytes) in dogs | No screening test abnormalities; evidence of hypercoagulability such as high D-dimer and/or FDP, high fibrinogen concentration, and normal or low AT activity; trends towards worsening hemostatic dysfunction (e.g. decreasing platelet count and increasing clotting times, FDP & D-dimer, decreasing AT activity) |
Treatment of DIC
Affected animals should always be treated for their primary disease with the goal of breaking or at least limiting the DIC cycle. Supportive care aimed at alleviating metabolic/hemodynamic sequelae of DIC (shock, hypoperfusion and acidosis) helps minimize organ damage, inflammation, and continued activation of hemostasis. Effective treatment modalities beyond primary disease-specific and supportive care remain unproven. General treatment options include transfusion therapy and anticoagulant drug therapy. Treatment recommendations are generally derived from the human literature and should only be used with the knowledge that they may not be suitable for animals, due to species-specific differences in hemostasis and drug pharmacokinetics and efficacy (i.e. they may hurt). Randomized, controlled clinical trials on DIC treatment are needed in veterinary medicine to identify safe and cost-effective DIC treatment strategies.
- Transfusion therapy: The fear that transfusions “fuel the fire” of DIC is largely unfounded, however transfusion therapy is generally restricted to actively bleeding patients in human studies (Wada et al 2013). Component therapy, rather than whole blood, generally provides the most effective means of restoring adequate levels of factors or platelets to support hemostasis. Fresh frozen plasma provides all coagulation factors and platelet concentrates can provide platelets in an emergency situation (e.g. acutely bleeding dog).
- Anticoagulants: Although thrombosis frequently underlies the morbidity and mortality of DIC, anticoagulant therapy, particularly heparin therapy, has not been proven to be a consistently effective for treatment of overt DIC in people or animals. Anticoagulants, such as heparin, should used with caution for treating DIC in animals, with recommendations for unfractionated and low molecular weight heparin (LMWH) therapy being adopted from humans. Platelet inhibitors, such as aspirin or clopidogrel, are generally not used to treat DIC (platelets may be dysfunctional from high concentrations of FDPs). Other inhibitors, such as APC, have anticoagulant and anti-inflammatory effects, and provision of inhibitor concentrates should be beneficial in treating DIC. Randomized clinical trials have shown some benefit of some of these inhibitor concentrates or recombinant proteins, e.g. recombinant thrombomodulin (Gando review). However, inhibitor concentrates or recombinant proteins are unavailable in animals and can only be provided from blood components. Since inflammation drives hemostasis activation, finding suitable anti-inflammatory drugs to treat affected animals would be beneficial (corticosteroids should be avoided as they may inhibit fibrinolysis and promote thrombosis; non-steroidal anti-inflammatory drugs, particularly COX-1 inhibitors, can inhibit platelet function).
DIC mimics
We rely on a constellation of laboratory tests to diagnose DIC and there are no diagnostic test results that are specific or unique for DIC. Observed changes in test results can be caused by other disorders or preanalytical variables without the patient being in DIC. For example, thrombocytopenia may be secondary to platelet clumping or mechanisms other than consumption (e.g. decreased production), a prolonged PT and APTT (the latter particularly) could be due to activation of coagulation factors with poor sample collection or over-citration of blood (e.g. underfilling the citrate tube), low AT could be secondary to protein-losing enteropathy or nephropathy, and high D-dimer could be due to neutrophil elastase-mediated cleavage of crosslinked fibrin or extravascular fibrinolysis (e.g. hemorrhage into a body cavity with D-dimer produced in the cavity being absorbed back into blood). Thus, it is important to interpret hemostatic test results in context of the case and make your best assessment of the status of the hemostatic system in light of clinical and other findings, not just the tests by themselves.
Two conditions that can mimic hemostatic changes (to some degree) seen in DIC are anticoagulant rodenticide toxicosis and liver failure, however there are differences in clinical presentation and laboratory test abnormalities (e.g. evidence of liver failure on a chemistry panel).
- Anticoagulant rodenticide toxicosis: Dogs can present with severe hemorrhage (often into body cavities), which may result in a thrombocytopenia (usually mild). The PT and APTT are usually both prolonged from decreased liver production of functional carboxylated vitamin K-dependent factors. Usually the PT is prolonged to a greater extent and/or earlier than the APTT, because FVII has the shortest half life of the coagulation factors. D-dimer can be increased due to extravascular fibrinolysis (breakdown of fibrin outside the vasculature), particularly with intracavity hemorrhage. Fibrinogen concentration is likely to be normal (not decreased as could be seen with consumption due to DIC and inflammation is not expected in animals with anticoagulant rodenticide toxicity). In addition, antithrombin concentrations are usually normal and, importantly, dogs do not have a primary illness that is causing DIC, i.e. their symptoms can be ascribed to hemorrhage rather than an underlying disease that can trigger a hemorrhagic manifestation of DIC.
- Liver failure: It can be very difficult, even in human medicine, to determine if abnormal hemostatic test results in animals with evidence of liver failure are due to the liver failure per se or concurrent DIC. DIC can be be concurrent with some conditions causing liver failure, e.g. hepatic toxins or infectious agents (e.g. leptospirosis), via the release of hepatocyte tissue factor or DAMPs from massive hepatic necrosis. In general, the platelet count is usually normal in liver failure in animals, whereas the count is usually low in DIC, particularly in the overt phase (there are always exceptions to these “rules”; platelet counts are less reliably decreased in DIC in other species, particularly cats). The PT and APTT may be prolonged due to decreased synthesis of coagulation factors as a direct consequence of liver failure or secondary to vitamin K deficiency associated with defective function of carboxylase enzymes or concurrent cholestasis (vitamin K is a fat soluble enzyme and requires bile acids for absorption; cholestasis or liver dysfunction can still prolong clotting times in a vitamin K-dependent manner in animals without liver failure). The PT is usually more prolonged with the APTT with synthetic failure or vitamin K-associated defects. Fibrinogen, AT and PC concentrations may be decreased due to synthetic failure. D-dimer, which is presumably cleared by Kupffer cells, may be increased if Kupffer cell function is also compromised. Thus, hemostatic test abnormalities in liver failure may mimic changes seen with overt DIC. Other clinical pathologic findings supportive of liver failure, such as hypocholesterolemia (most common), hypoalbuminemia, hypoglycemia and low urea nitrogen concentrations, may help identify liver failure as a cause of abnormal hemostasis results versus DIC in patients with evidence of liver disease (with the caveat above of concurrent DIC). A progressively falling platelet count and decreasing fibrinogen concentration may support DIC in animals with liver failure.