
Introduction
Definition: Primary hemostasis is defined as the formation of the primary platelet plug. This serves to plug off small injuries especially in microvessels (< 100 μm) in mucosal tissues (respiratory, gastrointestinal, genitourinary tracts). Platelets are not only involved in platelet plug formation but are also crucial for formation of fibrin (secondary hemostasis). Activated platelets express a negatively charged phospholipid, phosphatidylserine (PS), on their surfaces, which is a binding site for the assembly of coagulation factor complexes. Assembly of these complexes on this surface markedly amplifies fibrin formation (called the propagation phase of secondary hemostasis). In addition, activated platelets also activate neutrophils, leading to NETosis or extrusion of nuclear material (Carestia et al 2016), which is strongly procoagulant and antifibrinolytic (Liaw et al 2015).
Constituents: Platelets, von Willebrand factor (vWf), integrins, and extracellular matrix components in the vessel wall.
- Cells: Endothelial cells (source of vWf), platelets
- Adhesive proteins: vWf, integrin and non-integrin receptors, subendothelial matrix proteins (e.g. collagen)
- Facilitators: Thrombin (platelet agonist)

Sequence of events: Normally, the intact endothelium is a physical barrier separating circulating platelets from thrombogenic substances (such as extracellular matrix proteins) in the extravascular space. When the endothelium is injured, the procoagulant subendothelial matrix (consisting of proteins such as collagen, laminin, and fibronectin) is exposed and immediately initiates primary hemostasis, which consists of these events:
- Platelet capture: This occurs in vessels with high blood flow (shear rates) and is mediated by long strands of vWf, which capture and slow down circulating platelets.
- Platelet adhesion: Platelets firmly adhere to the exposed subendothelial matrix (directly to collagen or are captured via multimeric vWf).
- Platelet activation: Once platelets adhere, they then become activated and recruit (and activate) additional platelets to the injured site. Also, thrombin generated by the coagulation cascade is an extremely powerful platelet activator.
- Platelet plug formation: Fibrinogen forms bridges between activated platelets to form the platelet plug.
More detail on these three events is given below.
Note: Secondary hemostasis (formation of fibrin by coagulation factors) is usually initiated simultaneously (through exposure of tissue factor on subendothelial fibroblasts). Activated platelets promote fibrin formation and provide a physical scaffold on which fibrin formation proceeds. Thrombin generated by secondary hemostasis is a platelet agonist and also induces endothelial cells to release vWf from intracellular stores in Weibel-Palade bodies.
Sequence of events
Platelet capture and adhesion
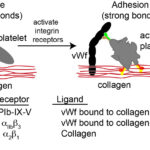
Upon endothelial injury, platelets bind to exposed subendothelial matrix proteins, such as collagen, with the aid of adhesion molecules or receptors on their surfaces. These receptors are transmembrane glycoproteins (GP) and are non-integrin or integrin receptors. Which receptors are involved in adhesion depends on the flow forces in the vessels. At higher shear rates, e.g. arterioles, platelets need to be slowed before they can firmly adhere, which is mediated by strands of vWf that unfurl from the subendothelial matrix or endothelial cell surface and allow platelets to transiently bind and roll, giving time for firm adhesion to occur via integrins. At lower shear rates, vWf is not required and firm binding can be mediated by the slower binding reactions of integrins on platelets with their ligands in the extracellular matrix. The main receptors involved in platelet capture/rolling and firm adhesion are the following:
- GP1b-IX-V: This hetero-tetrameric receptor is constitutively expressed on platelets and is found as a complex of two glycoproteins GP1b (with two subunits, alpha and beta) and GPIX in a ratio of 1:2:1 (Ibα:Ibβ:IX) (Quach and Li 2020). These two proteins bind weakly to GPV in a 1:1 ratio. The main ligand for the receptor is vWf, which as a long multimeric protein, acts as a long tether, rapidly capturing and slowing the velocity of circulating platelets to sites of vessel injury, by bridging extracellular matrix proteins and the receptor complex of GP1b-IX-V on the platelet surface. v
- Wf is produced by endothelial cells and stored in Weibel-Palade bodies. It is secreted constitutively into the subendothelial matrix and plasma (thus it can be measured in plasma). Even though vWf is in plasma, it does not bind to GP1b-IX-V (being in the incorrect compact conformation, which hides the receptor binding site); rather at high shear rates, collagen-tethered vWf is needed to capture platelets.
- With vessel injury, the multimeric vWf in the extracellular matrix unfurls into long strands (called “strings”) once exposed to the shear forces of blood and helps capture circulating platelets. Certain agonists, such as thrombin and histamine, can cause release of Weibel-Palade bodies and their stores of vWf from endothelial cells. The vWf remains tethered to the endothelial cell or is secreted into the subendothelial matrix, where both forms can also produce strings when exposed to flowing blood. Consisting of ultra-high molecular weight multimers, vWf from Weibel-Palade bodies contains many binding sites for capturing and slowing platelets.
- Once bound to vWf, GPIb-IX-V undergoes a conformational change, which triggers intracellular signaling and platelet activation (outside-in signaling) that activates the integrin-based receptors, such as GPIIb/IIIa (αIIbβ3) and GPIa/IIa (α2β1) on the platelet surface (Varga-Szabo et al 2008, Quach and Li 2020). The latter receptors, which have slow binding reactions, mediate firm adhesion to collagen (directly in the case of GPIa/IIa and indirectly via binding to vWf in the case of GPIIb/IIIa), whereas vWf-GPIb-IX-V bonds are weaker and more transient – like an “on-off” switch (Kuijpers et al 2004, Estevez and Du 2017). Thus, GPIb-IX-V/vWf interactions are important for mediating initial platelet interactions with vessels with high shear rates (as well as rolling of platelets), slowing down the platelets, so they can then bind firmly via the slower integrin-mediated reactions.
- Abnormalities in either GP1b-IX-V complex or vWf can result in a hemorrhagic diathesis. These are usually inherited disorders known as Bernard-Soulier syndrome (GP1b deficiency) or von Willebrand disease (vWD; abnormality or deficiency in vWf). Of these conditions, only vWD has been reported in animals.
- Integrins – α2β1and αIIbβ3: Once activated (usually via vWf binding to GPIb-IX-V), these integrins bind platelets firmly to collagen. The α2β1 integrin binds directly to collagen, whereas the fibrinogen receptor, αIIbβ3, binds to collagen-linked vWf, although it may also attach directly to specific amino acid residues (arginine-glycine-asparate; RGD) in other extracellular matrix proteins (fibronectin and vitronectin). GPIIb/IIIa also mediates platelet spreading on the vessel wall. β1-integrin-collagen interactions may be required for the full activation of GPIIb/IIIa (Nakamura et al 1999). Other β1-based integrin receptors may also help facilitate adhesion, such as fibronectin and laminin receptors. At low shear rates, integrin-mediated binding is often sufficient to yield platelet adhesion, without the requirement for initial slowing of platelet velocity by vWf-GPIb-IX-V interactions. Considering the importance of platelet adhesion in the cessation of bleeding, there is functional redundancy between receptors, with deficiencies in a single integrin reducing but not abolishing adhesion (Varga-Szabo et al 2008, Li et al 2010, Estevez and Du 2017).
Platelet activation
The adhesion of platelets to subendothelial matrix proteins activates the platelets (increasing intracellular calcium and inducing cell signaling) and causes a variety of changes in platelets. Binding of the non-integrin membrane protein, GPVI, optimizes collagen-induced platelet activation after adhesion has occurred (Kuijpers et al 2004, Estevez and Du 2017).
- Shape change: Platelets change from their normal discoid shape to elongated cells with cytoplasmic extensions, which increases their surface area.
- The release reaction (degranulation): Platelets release the contents of preformed cytoplasmic granules (α-granules and dense or δ-bodies).
- α-granules contain vWf and coagulation factors, including fibrinogen, Factor V (FV) and FXIII. Once released, these factors can participate in platelet plug formation (vWf, fibrinogen) or fibrin formation (FV, FXIII). Of these factors, only vWf is produced in megakaryocytes; the remaining proteins are endocytosed from plasma. There are also species differences in the amount of vWf within platelets, with dog platelets having little vWf, whereas cat platelets can contain up to 20% vWf. These granules also contain adhesion molecules, such as P-selectin, which is expressed on the platelet surface after activation. Detection of P selectin is actually used as an in vitro marker of platelet activation in the laboratory. In vivo, P selectin binds to its ligand, PSGL-1, on endothelial cells and leukocytes. This helps firmly platelets bind to the endothelium and allows leukocytes to be incorporated into developing clots (leukocytes, like platelets, also provide a surface scaffold for fibrin formation).
- Dense granules are rich in ADP and serotonin, both of which are platelet agonists (i.e. they recruit and activate additional platelets). They store calcium which is needed for secondary hemostasis and platelet activation. Recent studies have shown that they are also stores for short-chain polyphosphate (chains of phosphate molecules). These polyphosphates have been shown to have multiple roles in hemostasis and are both procoagulant (they promote the cofactor activity of Factor V in the common pathway and also serve as a cofactor for thrombin activation of FXI in the amplification phase of secondary hemostasis) and antifibrinolytic, i.e. they help form dense fibrin fibrils which are more resistant to fibrinolysis. Dense granule secretion can be measured by detecting the amount of 14C-serotonin or ATP released during aggregation.
- Note that the release reaction (via platelet factor IV, von Willebrand factor and P-selectin) also promote NET formation (release of nuclear material) from neutrophils (Carestia et al 2016), which are enmeshed in the developing clot, which is procoagulant and antifibrinolytic (Liaw et al 2015), providing a link between primary and secondary hemostasis.
- Phospholipid metabolism (membrane metabolism): Activation of phospholipase A2 hydrolyzes membrane phospholipids (phosphatidylcholine) to arachadonic acid. This is the initial substrate for subsequent reactions involving several enzymes (e.g. cyclo-oxygenase, COX-1) which produce eicosanoids, particularly thromboxane A2, which are powerful platelet agonists. Pharmacologic inhibition of COX-1 can be accomplished by aspirin and non-steroidal anti-inflammatory agents. These inhibitors are used to prevent thrombotic events in both people and animals (principally dogs).
- Phosphatidylserine exposure (membrane flipping): The normal platelet membrane is asymmetrical with the more negatively charged phospholipid, phosphatidylserine (PS), being preferentially and actively maintained on the inner membrane leaflet. Phosphatidylserine is very important, because it is the membrane surface on which the coagulation cascade occurs, i.e. it is the binding site for coagulation factors and actually amplifies their activity, thus promoting fibrin formation. When platelets are activated, PS is flipped to the outside of the platelet membrane, allowing coagulation factors to bind to the activated platelet. Phosphatidylserine used to be called platelet factor 3 (PF3). An inherited defect in PS exposure (Scott syndrome) has been reported in German Shepherd dogs and results in excessive hemorrhage in affected dogs. This has been shown to be due to a genetic defect in a calcium channel called TMEM16F in platelets, which is required for membrane flipping (Brooks et al 2002, Brooks et al 2016).
- Release of procoagulant microparticles (microvesiculation): Activated platelets shed tiny microparticles, that are enriched in PS, from their membrane surfaces. These microparticles extend the surface area of the platelet, thus providing a larger surface on which fibrin formation can proceed. Dogs with Scott syndrome also have a defect in microvesiculation. Phosphatidylserine exposure and microvesiculation can be assessed in the laboratory with flow cytometry (Brooks et al 2002).
- Recruitment of additional platelets (outside-in signaling): Additional platelets are recruited to the site of vessel injury by agonists that are released by activated platelets themselves (ADP, thromboxane A2) or that are formed from activated coagulation factors (thrombin). These agonists bind to surface receptors on platelets and trigger outside-in signaling which activates the recruited platelets. This outside-in signaling involves G proteins and other facilitator molecules, such as guanine exchange factors (GEF). An important GEF is CalDAG-GEF1 (calcium diacylglycerol-regulated guanine exchange factor-1) which is downstream of the ADP receptor and has shown to be defective in Basset Hounds and Eskimo Spitz with thrombopathia. Important platelet receptors for agonists are:
- P2Y12: This is the receptor for ADP. Clopidogrel is a P2Y12 antagonist that is also used as an antithrombotic drugs in humans, dogs and cats. Efficacy in horses is less certain.
- Protease-activated receptors (PAR): They are unique G-protein coupled receptors expressed on several different cells. They carry their own ligand, which is only expressed after the receptor is cleaved to expose the ligand, which then activates the receptor. Once activated, the receptor cannot be re-activated even though the cleavage enzyme dissociates from the receptor. These receptors bind several agonists, but the main agonist is thrombin, which binds to PAR-1 and PAR-4 expressed on human platelets, with PAR-1 being the main activator.
- Activation of the fibrinogen receptor (inside-out signaling): The integrin, αIIbβ3, also known as GPIIb/IIIa or CD41/61, is the fibrinogen receptor on platelets. It is normally present on platelet surfaces (indeed it is a platelet-specific marker) but cannot bind fibrinogen on unstimulated platelets. Once platelets are activated, the fibrinogen receptor changes conformation on the outside of the platelet due to intracellular signaling (inside-out), which allows fibrinogen to bind adjacent platelets, forming platelet aggregates. Binding of fibrinogen to activated platelets can be used as a laboratory marker of platelet activation. A genetic defect in GPIIb/IIIa has been detected in various breeds of dogs (Otterhound, Great Pyrenees) and cattle. This results in a bleeding disorder called Glanzmann’s thrombasthenia. A protein called kindlin is required for correct functioning of integrins in platelets and there has been a report of a dog with kindlin deficiency. The dog had combined leukocytosis, due to a leukocyte adhesion deficiency (integrins are required for leukocytes to migrate into tissue), and hemorrhagic defect due to defective platelet aggregation (Boudreaux et al 2010).
Platelet aggregation
This is mediated primarily by fibrinogen, which binds to the activated fibrinogen receptor (GPIIb/IIIa) on platelets. This links platelets together forming the primary plug. The platelet plug is sufficient to stop bleeding from most small blood vessels in response to every day trauma (e.g. eating) or venipuncture, however in larger vessels or with more severe damage, the primary platelet plug must be stabilized to cease the hemorrhage. This is accomplished through secondary hemostasis or fibrin production by coagulation factors. Also note, that platelets have roles beyond what has been described for hemostasis. For instance, it is now known that platelets are essential for vascular integrity. It is possible that hemorrhage in patients with thrombocytopenia is not only due to an absence of platelets but also to a loss of vascular integrity.
Platelet heterogeneity
It is now known that platelets are not a homogeneous population. There are subsets of platelets that are either procoagulant (express PS, assemble coagulation factor complexes and propagate thrombin generation) or those that are adhesive and contractile and more likely to bind to extracellular matrix proteins and cause clot retraction (Agbani and Pool 2017). Even within the procoagulant category, there are subtypes of different platelets, such as ballooned or “coated” platelets, the latter of which are formed from the dual activation of collagen and thrombin and have serotonin-conjugated proteins on their surface, including fibrinogen and thrombospondin (Dale 2005). Procoagulant platelets require robust sustained internal calcium-mediated signaling and strong agonists, such as collagen which binds to the collagen receptor, GPVI, or thrombin, which binds to PARs. Calcium signaling activates calpain and chloride entry via the chloride channel, TMEM16F (which is defective in Scott syndrome), which are required for microvesiculation and irreversible ballooning, and PS exposure (Agbani and Pool 2017). Platelet subsets are also located in different parts of the developing thrombus, with the adhesive platelets being found closer to the vessel wall and the procoagulant platelets forming an overlying cap.
Inhibitors
There are physiologic, pathologic and pharmacologic inhibitors of primary hemostasis.
- Physiologic: Endothelial cells release prostacyclin (PGE12) and nitric oxide, both of which inhibit platelet activation. They also form a physical barrier, preventing platelets from being exposed to thrombogenic subendothelial matrix proteins.
- Pathologic: High concentrations of fibrin(ogen) degradation products can inhibit platelet aggregation (fragments D and E have a high affinity for platelet membranes and compete with fibrinogen for platelet receptors, thus impairing aggregation) as can paraproteins (high concentrations of monoclonal immunoglobulins with multiple myeloma – the monoclonal protein coats platelets, interfering with platelet aggregation, adhesion and phospholipid exposure).
- Pharmacologic: These inhibit the following platelet activation events. Note: Drugs that inhibit platelet function can precipitate or worsen hemorrhage in an animal with a mild hemostatic disorder.
- Arachidonic acid metabolism: Aspirin (irreversibly) and non-steroidal anti-inflammatory drugs (reversibly) inhibit cyclo-oxgenase, which is needed for thromboxane A2 production.
- ADP agonist activity: Clopidogrel is an ADP receptor antagonist.
Clinical signs
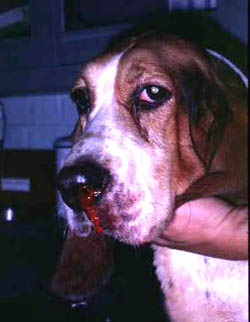
A defect in any aspect of primary hemostasis can manifest as hemorrhage, typically from mucosal surfaces (the sites where the primary platelet plug is important due to rapid fibrinolysis). Hyperactivity in primary hemostatic components is a cause of hypercoagulability and a risk factor for thrombosis.
- Defective primary hemostasis: Platelet plug formation is inadequate with a defect in any primary hemostasis event, including thrombocytopenia. This usually manifests as hemorrhage from small blood vessels subjected to daily minor trauma, i.e. those supplying mucuosa (e.g. epistaxis, hematuria). Hemorrhage can be spontaneous with severe defects or induced by trauma or surgery with milder defects. Since platelets plug small holes in vessels, thrombocytopenia or abnormal platelet function (thrombopathia) can result in pinpoint mucosal or cutaneous hemorrhages (petechiae). Petechiae are not typical in vWD. Bruises (ecchymoses) can also occur.
- Hyperactive primary hemostasis: Platelet hyper-reactivity and increased vWf concentrations contribute to hypercoagulability, i.e. are risk factors for thrombosis, but are unlikely to induce thrombosis alone. Thrombocytosis in animals is not usually associated with abnormal thrombotic events.
Sample collection
Two samples are required to evaluate primary hemostasis:
- EDTA-anticoagulated blood: Platelet count, genetic testing (if needed).
- Citrate-anticoagulated plasma: All other tests.
Please refer to the sample collection page for additional guidelines on how to collect samples appropriately to optimize hemostasis test results.
Tests
It is imperative that a platelet count is done in all animals presenting with clinical signs of excessive hemorrhage. Platelet counts can be quantified using automated analyzers or can be semi-quantified (estimated) from a well-prepared fresh blood smear. Additional platelet testing requires fresh blood or, preferably, referral of the patient to a testing center that is capable of performing advanced hemostasis testing. Few tests of primary hemostasis are available to general practitioners. These include a platelet count, a buccal mucosal bleeding time (BMBT), genetic testing for specific inherited disorders, and measurement of vWf concentrations.
- Screening tests: Platelet count, BMBT, platelet function analyzer testing.
- Specific tests: von Willebrand factor antigen (vWf:Ag), genetic testing for vWD and defined inherited thrombopathias (e.g. CalDAG-GEF1 defect in Bassett Hounds and Eskimo Spitz, GPIIb/IIIa deficiency or Glanzman’s thrombasthaenia in Otterhounds and Great Pyrenees), TMEM16F genetic defect in Scott Syndrome (Brooks et al 2016).
- Specialized testing: Flow cytometric tests for platelet activation (P selectin expression), function in primary hemostasis (platelet aggregation, release reaction), procoagulant activity (PS exteriorization, microparticle quantification, thrombin generation assays), platelet-associated antibodies, reticulated platelets.
Disorders
Defects arise in any aspect of the hemostatic pathway. As indicated above, these defects can result in deficient hemostasis (hypocoagulability), which can result in excessive hemorrhage, or accelerated hemostasis (hypercoagulability), which can result in thrombosis. Disorders can also be inherited or acquired. Inherited conditions should be suspected in young animals presenting with episodes of recurrent hemorrhage or thrombosis. Acquired disorders are more likely in older animals with underlying disease. The most common inherited defect in primary hemostasis is von Willebrand Disease and thrombocytopenia is the most common acquired defect (there are multiple causes of thrombocytopenia).