
Introduction
Definition: Secondary hemostasis is defined as the formation of insoluble, cross-linked fibrin by activated coagulation factors, specifically thrombin. Fibrin stabilizes the primary platelet plug, particularly in larger blood vessels where the platelet plug is insufficient alone to stop hemorrhage.
Synonym: Coagulation
Constituents: These consist of cells, enzymatic and non-enzymatic coagulation factors, protein substrates, calcium and phospholipid (phosphatidylserine, PS) membranes. Coagulation factors can be proteolytic enzymes (zymogens) or non-enzymatic. Non-enzymatic coagulation factors can be cofactors for enzymatic coagulation factors or can just be a substrate (Factor I or fibrinogen). Enzymatic and non-enzymatic coagulation factors are substrates for enzymatic coagulation factors. Phosphatidylserine-bearing cell membranes are usually provided by activated platelets, but leukocytes and erythrocytes, which are incorporated into the developing clot, can also expose PS on their cell surfaces and be a source of PS.
- Cells: Fibroblasts, platelets, endothelial cells, leukocytes
- Enzymatic coagulation factors: Factors XI, X, IX, VII, and II. These are usually in an inactive form and must be activated before they can exert their enzymatic (cleavage) activity.
- Non-enzymatic coagulation factors – cofactors: Tissue factor (TF), Factors V and VIII. Both Factor V and Factor VIII, but not tissue factor, require activation (by an enzymatic coagulation factor, usually thrombin or Factor IIa).
- Non-enzymatic coagulation factor – substrate: Fibrinogen
- Calcium: Required for all steps in secondary hemostasis
- Phosphatidylserine (PS)-bearing membrane surfaces.
Nomenclature: Coagulation factors are identified as roman numerals, based on the order in which they were isolated and identified historically, e.g. fibrinogen is also coagulation factor I (FI). Coagulation factors (zymogens and cofactors) are present in plasma as unactivated and activated proteins. By convention, the activated form of the coagulation factor is denoted by a small “a” after the factor number, e.g. FVII is the proenzyme and FVIIa is the active enzyme.
Coagulation pathways
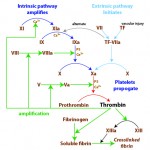
Traditionally, coagulation has been divided into three distinct pathways: extrinsic, intrinsic and common. The constituents of each pathway are shown below.
- Extrinsic pathway: This is composed of:
- Tissue factor (cofactor) and FVII (pro-enzyme; FVIIa is the enzyme)
- Calcium (Ca2+)
- The TF-FVIIa complex activates FX of the common pathway; it is thus called the “extrinsic tenase” (extrinsic pathway Factor X activator). The TF-FVIIa complex can also activate FIX of the intrinsic pathway (in the so-called “alternate pathway”), although FX is its preferred substrate in vitro. Under physiologic conditions (blood vessel injury), FXa is generated by the TF-FVIIa complex on the surface of fibroblasts.
- Intrinsic pathway: This is composed of:
- Enzymatic coagulation factors: FXII, FXI, and FIX
- The cofactor (non-enzyme): FVIII
- Ca2+
- PS
- The ultimate product of the intrinsic pathway is activated FIX, which (with the aid of activated cofactor FVIIIa), activates FX. In fact, the FIX-FVIIIa-Ca2+-PS complex is called the “intrinsic tenase” (intrinsic pathway Factor X activator). The FXa is generated by the intrinsic tenase on the platelet surface. Thus both intrinsic and extrinsic pathways converge at the activation of FX, however the site of activation of FX differs (fibroblast for the extrinsic pathway and platelet for the intrinsic pathway).
- Common pathway: This is composed of:
- Enzymatic coagulation factors: FX, prothrombin (FII) and FXIII (crosslinker)
- The cofactor: FV
- Protein substrate: Fibrinogen
- Ca2+
- PS
- FXa, with the aid of its cofactor FVa, activates prothrombin to thrombin. Thrombin activates FXIII and cleaves fibrinogen to soluble fibrin, which is then crosslinked to insoluble fibrin by FXIIIa.
These pathways interact on the surface of cells to generate thrombin, which cleaves and crosslinks fibrin.
Sequence of events
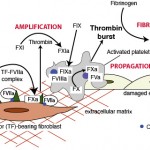
Secondary hemostasis (coagulation) is usually initiated simultaneously with primary hemostasis upon endothelial damage, although this depends on the degree and location of injury. Secondary hemostasis consists of a sequence of events:
- Initiation of thrombin generation: This occurs through tissue factor (extrinsic pathway), which is expressed on fibroblasts. This generates small amounts of thrombin and Factor IXa.
- Amplification of thrombin generation: This occurs through thrombin, which activates Factor XI of the intrinsic pathway (with platelet polyphosphates as a cofactor) and the cofactors Factor VIII (intrinsic) and Factor V (common). This phase proceeds on activated platelets (thrombin is a strong platelet agonist) and potentially other cell surfaces expressing phosphatidylserine (endothelial cells, white blood cells, red blood cells), particularly in platelet-poor parts of a growing clot. This generates a large amount of Factor Xa on the platelet surface.
- Propagation of thrombin generation: This occurs through Factor Xa on the platelet surface, with the help of the cofactor, Factor Va, and generates an explosive thrombin burst.
- Fibrin formation: The explosive thrombin burst cleaves fibrinogen to fibrin, which then forms a soluble fibrin polymer and network of polymers. This network is stabilized by Factor XIII, which crosslinks the fibrin. Thrombin is responsible for activating Factor XIII and simultaneously inhibiting fibrinolysis, by activating thrombin-activatable fibrinolysis inhibitor. Polyphosphates released from dense granules of activated platelets also inhibit fibrinolysis by helping form a dense fibrin network.
Thrombin generation occurs on the PS-bearing surface of cells, particularly platelets (per current dogma), although leukocytes are likely involved as are endothelial cells. The PS-bearing membrane surface is essential for optimal fibrin formation – it acts as physical scaffold for coagulation, provides binding sites for the assembled coagulation factor complexes, markedly enhances coagulation factor complex activity (by 1000 fold) and protects activated coagulation factors from inhibitors. Platelets not only provide PS surfaces that support and propagate fibrin formation but they also produce high local concentrations of coagulation factors (FV, fibrinogen, FXIII) which participate in clot formation. They also release procoagulant short-chain polyphosphates which promote amplify thrombin generation, promote fibrin formation and inhibit fibrinolysis (by forming a dense fibrin clot). Note, that there is no role for factor XII in activation of physiologic hemostasis (this is why factor XII-deficient humans and animals do not bleed). Factor XII, which is now designated as part of the contact pathway of coagulation (with high molecular weight kininogen and prekallikrein) has other physiologic roles, including activation of fibrinolysis, angiogenesis and inflammation (Woodruff et al., 2011). Although FXII is not needed for physiologic hemostasis, there is ample evidence from animal models that FXII is involved in pathologic thrombosis, since factor XII deficiency protects animals from thrombosis in experimental models (Renne and Gailani, 2007).
Note: In the past, both extrinsic pathway and intrinsic pathways of coagulation were thought to be important in initiation of coagulation – this was called the waterfall or cascade model of coagulation and led to the term “the coagulation cascade”. The intrinsic pathway was thought to initiate secondary hemostasis through FXII, which is activated by contact with negatively charged surfaces, such as glass. Activation of both pathways simultaneously led to a cascade or waterfall effect, with inactive enzymes becoming activated in a sequential manner, and each subsequent step being amplified. The two pathways converged where FX is converted to FXa, with the downstream reactions called the common pathway. However, this waterfall model is quite simplified and does not take into account inter-relationships between coagulation factors within the cascade (e.g. thrombin activates FXI, FVIII and FV) and between coagulation factors and negatively charged cell surfaces. Also, it is now known that FXII of the intrinsic pathway has no role in the initiation of physiologic hemostasis (recent studies in mice indicate that factor XII may, however, be involved in pathologic thrombosis). Indeed, some animals (aquatic mammals, birds, some cats) are deficient in FXII and do not bleed excessively. FXII is thought to be involved in fibrinolysis amongst other things. The waterfall model has been replaced by the cell-based model, which as described in more detail belos.
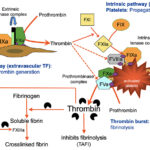
Initiation of thrombin generation
This is accomplished by the extrinsic pathway and involves the following players:
- Cell: Fibroblast
- Enzymes: Factors VII, X, IX
- Cofactors: Tissue factor (Factor III), calcium, PS
Initiation is mediated via TF, which is constitutively expressed in perivascular fibroblasts and is a transmembrane protein with no intrinsic enzymatic activity. Endothelial injury exposes TF on fibroblasts to its circulating ligand in plasma, the pro-enzyme FVII (small amounts of FVII are found in the activated state, FVIIa, in plasma – around 1% ; this FVIIa cannot initiate coagulation because it lacks the cofactor activity of tissue factor, which is isolated from circulating FVIIa by an intact endothelial surface). Binding to TF activates FVII and promotes its activity. The TF-FVIIa complex then activates the proenzyme FX. FXa then activates its cofactor FV (slowly) and the “extrinsic tenase” of FXa-FVa-PS-Ca2+ generates a small amount of the enzyme thrombin. This small amount of thrombin is insufficient to form fibrin but can amplify coagulation and activate platelets, which are essential for the amplification and propagation of thrombin generation. The TF-VIIa complex can also activate FIX directly, which is called “the alternate” pathway. This FIXa can participate in the amplification phase of thrombin generation. Thus, initiation occurs on the surface of fibroblasts and the end-point is generation of small amounts of thrombin (which then amplifies its own production) and FIXa, which can diffuse off the fibroblast surface onto the platelet surface to help amplify and propogate thrombin generation. Unlike FIXa, FXa cannot effectively diffuse off the fibroblast surface to participate in propagation and amplification. This is because FXa is rapidly inhibited by tissue factor pathway inhibitor and antithrombin, when not surface bound.
Note, new evidence suggests that TF, even that in extravascular spaces below an intact endothelium, is bound to FVIIa and that the trigger for activation of hemostasis is exposure of the TF-FVIIa complex on fibroblasts to its substrate, factor X, which is too large to enter the extravascular space. This prebound TF-FVIIa has given arise to the concept of an “idling” hemostatic system, poised to get moving quickly when vessels are injured (Hoffman et al., 2007). In addition, phosphatidic acid [PA] and not phosphatidylserine may be the main ligand for surface binding of FVII, which actually binds weakly to phosphatidylserine (Tavoosi et al., 2013).
Amplification of thrombin generation
In this phase, coagulation moves from the fibroblast surface to the platelet surface and thrombin amplifies its own production, producing a large burst of thrombin that is essential for fibrin formation and inhibition of fibrinolysis. This amplification phase is accomplished via thrombin activating intrinsic pathway factors. Amplification involves the following players:
- Cells: Activated platelet (and likely endothelial cells and leukocytes).
- Enzymes: Factors II, FXI, FIXa
- Cofactors: FVIII, calcium, PS, polyphosphates
The small amount of thrombin generated by TF-FVIIa in the initiation phase then amplifies coagulation by activating FXI (with the help of platelet-released polyphosphates, which accelerate the reaction [Choi et al 2011]) and FVIII, the cofactor of FIXa. FXIa then amplifies the activation of FIX on the surface of the activated platelet and in the presence of calcium. Factor XIa has also been shown to cleave tissue factor pathway inhibitor (the physiological inhibitor of TF-FVIIa-FXa), which may promote FXa activation via the extrinsic pathway (Puy et al 2015). Factor IXa generated by the extrinsic factor complex of TF-FVIIa can diffuse from the fibroblast surface to the platelet surface, increasing the amount of FIXa that can be incorporated into the “intrinsic tenase” complex, which consists of FIXa, FVIIIa, PS and calcium. Studies have shown that FVIII binds to activated platelets (when stimulated by thrombin and other agonists such as collagen) via PS, although FVIII may bind to activated platelets (when stimulated by thrombin alone) via fibrin bound to GPIIa/IIIb (the fibrinogen receptor) (Gilbert et al 2015). FXI has also been shown to inactivate tissue factor pathway inhibitor, thereby enhancing TF-FVIIa-mediated thrombin generation (Puy et al 2015). FXIa can also activate FIX in a phospholipid-dependent manner (Mohammed et al 2018).
Propagation of thrombin generation
This is accomplished by phosphatidylserine-bearing cell membrane surfaces, the intrinsic tenase complex (FIX-FVIIIa-FX), and the prothrombinase complex (FXa-FVa-FII) and involves the following players:
- Cells expressing PS: Activated platelets are thought to be the primary surface, but there is increasing evidence that PS-expressing platelets are found in a focal area of a growing clot close to the vessel wall, whereas thrombin generation occurs in other areas containing fewer or less activated platelets. This suggests that other cells, not only platelets, support thrombin generation, such as endothelial cells and leukocytes (Ivanciu and Stalker 2015).
- Enzymes: Factors FIXa, FX, FII
- Cofactors: FVIII, FV, polyphosphates, calcium, PS
The “intrinsic tenase” complex of FIXa-FVIIIa–PS-Ca2+amplifies the activation of FX on the PS-expressing cell surface (defaulted to platelets). Activated FX forms a complex with its cofactor FVa (activated mostly by thrombin in the amplification event and released locally by platelets after they degranulate – once released FV binds to activated platelets via PS) and calcium on platelet PS surfaces. This “prothrombinase” complex then generates an explosive burst of thrombin from prothrombin. Thrombin-mediated platelet activation will increase PS exposure and release coagulation factors and cofactors from intracellular stores, resulting in localized high concentrations of FV and polyphosphates (right where they are needed).
Fibrin formation
This is accomplished by large amounts of thrombin which cleaves fibrinogen to fibrin and activates the enzyme that crosslinks fibrin. This involves the following players:
- Cells: Activated platelet (or other cells)
- Enzymes: Factors FIIa, FXIII
- Substrate: Fibrinogen
- Cofactors: PS, calcium, polyphosphate
- Facilitator: Thrombin-activatable fibrinolysis inhibitor (TAFI)
The large amount of thrombin generated through amplification and propagation produces the fibrin clot and simultaneously prevents its dissolution (inhibition of fibrinolysis). Fibrin is generated from fibrinogen in two steps by large amounts of thrombin. First, there is the formation of the soluble fibrin monomer, which polymerizes and laterally associates to form a soluble fibrin network. Second, there is the stabilization of this network via crosslinking, which is achieved by activated Factor XIII in the presence of ionized calcium. Fibrinogen consists of a duplex of α, β and γ chains. Each set of chains are linked together at one end, forming a molecule, consisting of two terminal ends (D domains) and a central (linked) E domain. Initially, the fibrinogen is cleaved at the E domain by thrombin to form soluble fibrin. This cleavage releases small peptides from the terminal ends of the α and β chains of fibrinogen, called fibrinopeptide A (from the α chain) and B (from the β chain). These peptides are used in humans as markers of thrombin generation. The cleavage of peptides (particularly fibrinopeptide A) allows soluble fibrin to spontaneously associate into long fibrils (fibrin polymer) by covalent associations between terminal (D) ends of each fibrin monomer. Each polymer is also associated loosely with adjacent polymers. The fibrin polymer network is then stabilized by the enzyme, FXIII, which is activated by thrombin. Factor XIIIa crosslinks the γ chains of the terminal (D) ends of each fibrin monomer within each polymer, creating the neo-epitope D-dimer in the process. Factor XIIIa also crosslinks adjacent fibrin polymers through their α chains, forming a three-dimensional crosslinked network. At the same time that thrombin is forming fibrin, it is also activating an inhibitor of fibrinolysis called thrombin-activatable fibrinolysis inhibitor or TAFI. Thus, as the fibrin clot is developing, fibrinolysis is inhibited preventing its breakdown. Fibrin incorporates within and stabilizes the primary platelet plug and is also important for binding the platelet plug to the injured vessel wall.
Factors affecting fibrin clot formation
The strength of the formed clot depends on several factors, including the amount and quality of fibrinogen, activity of Factor XIII, polyphosphates, extracellular DNA and the amount of thrombin generated. Large amounts of thrombin generated by amplification and propagation form thick dense fibrils, which are more resistant to fibrinolysis. In contrast, smaller amounts of thrombin form thinner and weaker fibrils, that can be broken down more readily. So it is not only the amount of fibrin formed that matters, it is the nature of the fibrin formed that is also important as this dictates the strength of the clot and how long it will last for.
- Polyphosphates released from activated platelets (stored in dense granules) help make a firmer denser clot, that resists fibrinolysis (i.e. they are antifibrinolytic).
- Extracellular DNA: Neutrophils are part of a thrombus and these cells can extrude their nuclei when activated (a process called NETosis), releasing DNA and citrullinated histones. This DNA is procoagulant (due to their negative charge, DNA promotes thrombin generation via contact activation and the intrinsic pathway) (Massberg et al 2010, Gould et al 2014) and the extracellular strands of DNA act like fibrin, helping to enmesh platelets within the clot. The cell-free DNA also is anti-fibrinolytic (binding to and inhibiting plasmin) (Gould et al 2014). Administration of DNaseI, which degrades DNA, protects mice from thrombosis in a model of deep vein thrombosis caused by almost complete blockage of the inferior vena cava (Brill et al 2012). Note that other cell types can also release extracellular DNA and histones and these cells along with NETosis by activated neutrophils may be involved in the pathogenesis of DIC (Liaw et al 2015) and pathologic thrombosis in other disease states, such as bacterial sepsis, cancer (Demers et al 2012) and IMHA in dogs.
Role of vitamin K
Vitamin K is an essential cofactor in the carboxylation of γ-glutamic acid (GLA) residues of the amino terminal domains of factors II, VII, IX and X, which occurs during synthesis of these proteins in the liver. The inhibitors, protein C and protein S, are also vitamin K-dependent. Carboxylation of the GLA-residues allows these coagulation factors to bind calcium, which is required for binding to PS on platelet surfaces. Non-carboxylated factors are “inactive” because they cannot bind calcium or to PS and, therefore, cannot participate in the formation of the essential coagulation complexes that are required for fibrin formation. Thus, fibrin formation is defective when vitamin K is deficient. Warfarin and warfarin-based anticoagulant rodenticides result in a relative vitamin K deficiency because they inhibit vitamin K epoxide reductase, which is necessary for recycling of vitamin K to its active form in the vitamin K-epoxide cycle. Vitamin K is also a fat-soluble enzyme (hence vitamin K deficiency can also occur in cholestatic disorders).
Inhibitors
There are physiologic, pharmacologic and pathologic inhibitors of secondary hemostasis.
- Physiologic:
- Extrinsic pathway: Tissue factor pathway inhibitor (TFPI) is the main inhibitor of this pathway. TFPI inhibits the TF-FVIIa complex and the TF-FVIIa-FXa complex. TFPI rapidly downregulates the initiation phase of coagulation, hence thrombin has to amplify its own production. Activity of TFPI is promoted by protein S, which is also a cofactor for an inhibitor of thrombin, protein C. Antithrombin also weakly inhibits FVIIa, with stronger inhibition occurring in the presence of heparin-like glycosaminoglycans.
- Intrinsic pathway: The main inhibitors are antithrombin (AT) and protein C.
- Antithrombin: AT is an α2-globulin that is produced in the liver. It is an important endogenous anticoagulant, inhibiting the activity of most activated coagulation factors (FIIa, IXa, Xa, XIa and XIIa), although its greatest inhibitory effect is against thrombin (FIIa). Its activity is enhanced by heparin (hence its use as a prophylactic anticoagulant for thrombosis and as an in vitro anticoagulant to provide plasma for laboratory testing), which is provided in vivo by heparin-like glycosyaminoglycans on the surface of endothelial cells. Antithrombin complexes with thrombin, the complex is then removed by the monocyte-macrophage system.
- Protein C: This is a thrombin inhibitor and vitamin K-dependent enzyme that is produced in the liver. Thus, it is antithrombotic and profibrinolytic. Protein C is activated when thrombin binds to thrombomodulin on endothelial cells (the endothelial protein C receptor works as a cofactor in this activation) and works by inhibiting the intrinsic tenase and prothrombinase cofactors, FVIIIa and FVa, respectively. Inhibition of these cofactors markedly decreases activity of the complexes, slowing thrombin generation. Activity of protein C is enhanced by free protein S, which is also vitamin K-dependent and produced in the liver.
- Protein Z: This is a factor X inhibitor.
- Heparin-like glycosaminoglycans: These are found on the surface of endothelial cells and boost the inhibitory activity of AT.
- Heparin cofactor II: This is a specific thrombin antagonist. Like AT, this also requires heparin for activation, but in far greater concentrations.
- Non-specific: These are protease inhibitors found in plasma, e.g.α1-antitrypsin, α2-macroglobulin. They non-specifically inhibit any proteolytic enzymes, including activated coagulation factor zymogens.
- Pathologic: High concentrations of fibrin degradation products can inhibit fibrin polymerization as can paraproteins (high concentrations of monoclonal immunoglobulins with multiple myeloma). Inhibitors of coagulation factors can arise in certain autoimmune diseases, e.g. antiphospholipid antibodies in systemic lupus erythematosis.
- Pharmacologic: This includes heparin (which potentiates the action of antithrombin) and warfarin, which inhibits recycling of vitamin K, resulting in a relative deficiency of vitamin K-dependent factors (FII, VII, IX, X) – these are not truly deficient (they are being produced); they just cannot bind to PS and become activated. These are used to prevent thrombosis in animals.
- Heparin: This is frequently administered as an anticoagulant in animals with hypercoagulability (e.g. immune-mediated hemolytic anemia in dogs). Both fractionated and unfractionated heparin act as anticoagulants via AT. The activity of heparin depends on the number of pentasaccharides. More pentasaccharides as found in unfractionated heparin inhibit both FXa and thrombin. In contrast, shorter chain heparin as found in low molecular weight or fractionated heparin only inhibit FXa. The latter have more predictable pharmacodynamics and are less likely to cause excessive bleeding. The inhibitory effect of heparin can be evaluated by prolongation of the APTT (unfractionated) or decreased FXa activity (unfractionated and low molecular weight heparin).
Clinical signs
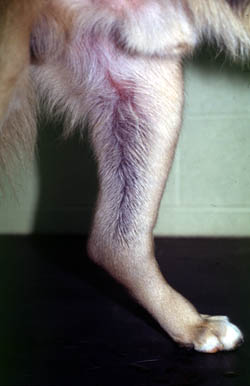
(Photograph courtesy of Dr. Marjory Brooks).
Defects in secondary hemostasis usually manifest as hemorrhage. Hyperactivity in secondary hemostatic components can be a risk factor for thrombosis, but is less commonly recognized.
- Deficiency or abnormal function of coagulation factors: Since fibrin is required to stabilize the platelet plug, decreased fibrin formation from coagulation factor or vitamin K deficiency results in bleeding from larger vessels. This manifests as large ecchymoses (hematomas) and hemorrhage into body cavities (hemarthrosis, hemoperitoneum etc). As for defects in primary hemostasis, traumatic- or surgical-induced hemorrhage can occur and this does not discriminate between abnormalities in primary or secondary hemostasis. Deficiencies of inhibitors can lead to thrombosis.
- Hypercoagulability: This is defined as excessive generation of thrombin and is caused by aberrant activation of secondary hemostasis, usually by TF (e.g. massive endothelial injury, aberrant intravascular expression of TF on monocytes or cancer cells) combined with a lack of adequate inhibition. Animals in hypercoagulable states are at risk of or may already be developing microvascular thrombi. High concentrations of some coagulation factors, such as fibrinogen and FVIII, may contribute to a hypercoagulable state, but alone will not result in thrombosis. Thrombosis is very difficult to recognize clinically. This is because thrombi frequently occur in internal small vessels, thrombotic sequelae of hypoxic injury to vital organs may be masked by other diseases, and there is a paucity of sensitive or specific diagnostic or imaging tests for thrombi.
Sample collection
The following samples are required for evaluation of secondary hemostasis:
- EDTA-anticoagulated blood or cheek swab: Genetic tests.
- ACT tube: Activated coagulation time. The tube contains a contact activator (activates factor XII), which varies depending on the manufacturer. The activator in standard ACT tubes was diatomaceous earth. However, these are not available through some supplier and newer tubes contain a different activator (e.g. a combination of celite, kaolin and glass beads) and ACT times are different and specific for activator.
- Citrate-anticoagulated plasma: All other assays.
Please refer to the sample collection section on more guidelines on how to collect samples appropriately to optimize coagulation test results.
Tests
- Screening tests: Prothrombin time (PT), activated partial thromboplastin time (APTT), fibrinogen concentration, activated coagulation time (ACT), proteins induced by vitamin K antagonism/absence (PIVKA)
- Specific tests: Individual coagulation factor activity assays, genetic tests for specific inherited disorders.
- Tests for inhibitors: AT and activated protein C assays, therapeutic heparin monitoring assays (anti-FXa activity), anticoagulant rodenticide testing.
- Specialized tests: Antithrombin-thrombin complexes (TAT), thrombin generation assays, pathologic coagulation factor inhibitors (e.g. antiphospholipid antibodies), fibrinogen antigen.
Disorders
Defects arise in any aspect of the hemostatic pathway. As indicated above, these defects can result in deficient hemostasis (hypocoagulability), which can result in excessive hemorrhage, or accelerated hemostasis (hypercoagulability), which can result in thrombosis. Disorders can also be inherited or acquired. Inherited conditions should be suspected in young animals presenting with episodes of recurrent hemorrhage or thrombosis. Acquired disorders are more likely in older animals with underlying disease. The most common inherited defect of secondary hemostasis is hemophilia A or FVIII deficiency and the most common acquired disorders are anticoagulant rodenticide toxicosis (causing vitamin K antagonism) and DIC.